BRCA2 and BRCA2 (/ˌbrækəˈtuː/[5]) are human genes and their protein products, respectively. The official symbol (BRCA2, italic for the gene, nonitalic for the protein) and the official name (originally breast cancer 2; currently BRCA2, DNA repair associated) are maintained by the HUGO Gene Nomenclature Committee. One alternative symbol, FANCD1, recognizes its association with the FANC protein complex. Orthologs, styled Brca2 and Brca2, are common in other vertebrate species.[6][7] BRCA2 is a human tumor suppressor gene[8][9] (specifically, a caretaker gene), found in all humans; its protein, also called by the synonym breast cancer type 2 susceptibility protein, is responsible for repairing DNA.[10]






BRCA2 and BRCA1 are normally expressed in the cells of breast and other tissue, where they help repair damaged DNA or destroy cells if DNA cannot be repaired. They are involved in the repair of chromosomal damage with an important role in the error-free repair of DNA double strand breaks.[11][12] If BRCA1 or BRCA2 itself is damaged by a BRCA mutation, damaged DNA is not repaired properly, and this increases the risk for breast cancer.[13][14] BRCA1 and BRCA2 have been described as "breast cancer susceptibility genes" and "breast cancer susceptibility proteins". The predominant allele has a normal tumor suppressive function whereas high penetrance mutations in these genes cause a loss of tumor suppressive function, which correlates with an increased risk of breast cancer.[15]
The BRCA2 gene is located on the long (q) arm of chromosome 13 at position 12.3 (13q12.3).[16] The human reference BRCA2 gene contains 27 exons, and the cDNA has 10,254 base pairs[17] coding for a protein of 3418 amino acids.[18][19]
Function edit

Although the structures of the BRCA1 and BRCA2 genes are very different, at least some functions are interrelated. The proteins made by both genes are essential for repairing damaged DNA (see Figure of recombinational repair steps). BRCA2 binds the single strand DNA and directly interacts with the recombinase RAD51 to stimulate[27] and maintain [28] strand invasion, a vital step of homologous recombination. The localization of RAD51 to the DNA double-strand break requires the formation of the BRCA1-PALB2-BRCA2 complex. PALB2 (Partner and localizer of BRCA2)[29] can function synergistically with a BRCA2 chimera (termed piccolo, or piBRCA2) to further promote strand invasion.[30] These breaks can be caused by natural and medical radiation or other environmental exposures, but also occur when chromosomes exchange genetic material during a special type of cell division that creates sperm and eggs (meiosis). Double strand breaks are also generated during repair of DNA cross links. By repairing DNA, these proteins play a role in maintaining the stability of the human genome and prevent dangerous gene rearrangements that can lead to hematologic and other cancers.
BRCA2 has been shown to possess a crucial role in protection from the MRE11-dependent nucleolytic degradation of the reversed forks that are forming during DNA replication fork stalling (caused by obstacles such as mutations, intercalating agents etc.).[31]
Like BRCA1, BRCA2 probably regulates the activity of other genes and plays a critical role in embryo development.
Clinical significance edit

Certain variations of the BRCA2 gene increase risks for breast cancer as part of a hereditary breast–ovarian cancer syndrome. Researchers have identified hundreds of mutations in the BRCA2 gene, many of which cause an increased risk of cancer. BRCA2 mutations are usually insertions or deletions of a small number of DNA base pairs in the gene. As a result of these mutations, the protein product of the BRCA2 gene is abnormal, and does not function properly. Researchers believe that the defective BRCA2 protein is unable to fix DNA damage that occurs throughout the genome. As a result, there is an increase in mutations due to error-prone translesion synthesis past un-repaired DNA damage, and some of these mutations can cause cells to divide in an uncontrolled way and form a tumor.
People who have two mutated copies of the BRCA2 gene have one type of Fanconi anemia. This condition is caused by extremely reduced levels of the BRCA2 protein in cells, which allows the accumulation of damaged DNA. Patients with Fanconi anemia are prone to several types of leukemia (a type of blood cell cancer); solid tumors, particularly of the head, neck, skin, and reproductive organs; and bone marrow suppression (reduced blood cell production that leads to anemia). Women having inherited a defective BRCA1 or BRCA2 gene have risks for breast and ovarian cancer that are so high and seem so selective that many mutation carriers choose to have prophylactic surgery. There has been much conjecture to explain such apparently striking tissue specificity. Major determinants of where BRCA1- and BRCA2-associated hereditary cancers occur are related to tissue specificity of the cancer pathogen, the agent that causes chronic inflammation, or the carcinogen. The target tissue may have receptors for the pathogen, become selectively exposed to carcinogens and an infectious process. An innate genomic deficit impairs normal responses and exacerbates the susceptibility to disease in organ targets. This theory also fits data for several tumor suppressors beyond BRCA1 or BRCA2. A major advantage of this model is that it suggests there are some options in addition to prophylactic surgery.[33]
In addition to breast cancer in men and women, mutations in BRCA2 also lead to an increased risk of ovarian, uterine tube, prostate and pancreatic cancer. In some studies, mutations in the central part of the gene have been associated with a higher risk of ovarian cancer and a lower risk of prostate cancer than mutations in other parts of the gene. Several other types of cancer have also been seen in certain families with BRCA2 mutations.
In general, strongly inherited gene mutations (including mutations in BRCA2) account for only 5-10% of breast cancer cases; the specific risk of getting breast or other cancer for anyone carrying a BRCA2 mutation depends on many factors.[34]
History edit
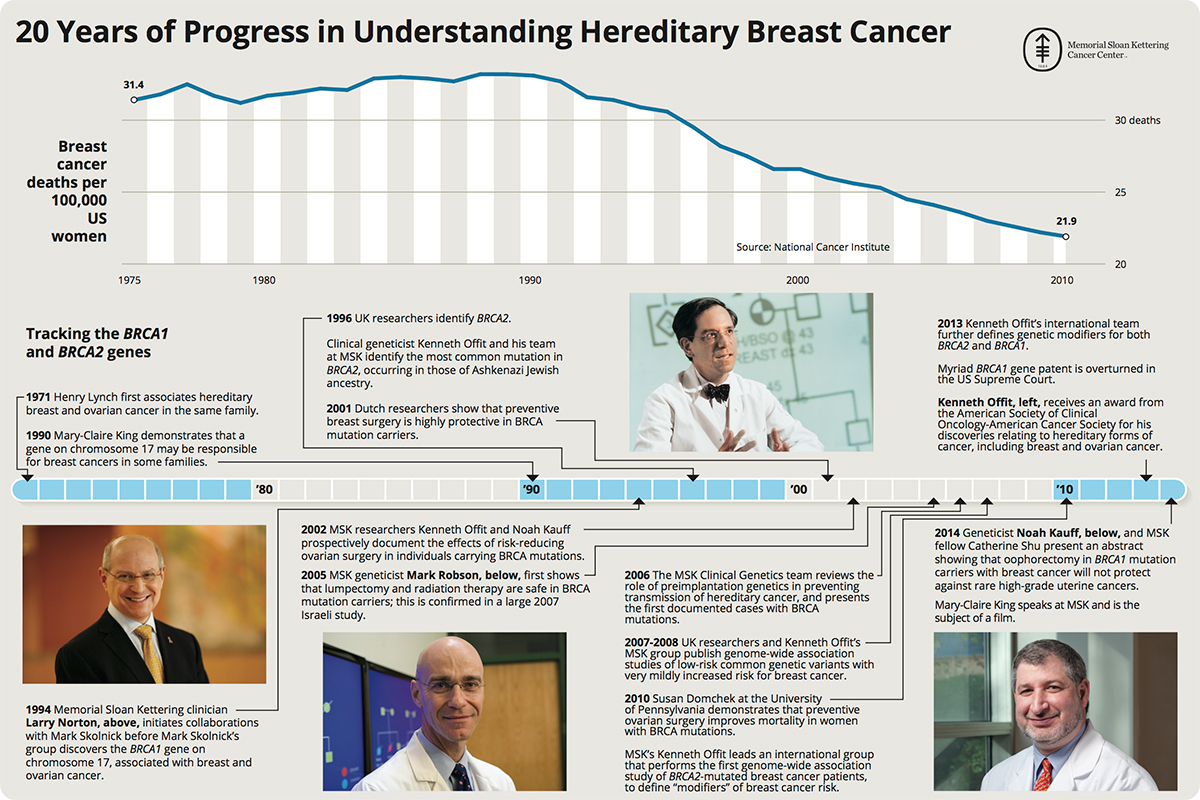
The BRCA2 gene was discovered in 1994.[35][16][36] In 1996, Kenneth Offit and his research group at Memorial Sloan Kettering Cancer Center successfully identified the most common mutation on the gene associated with breast and ovarian cancer among individuals of Ashkenazi Jewish ancestry.[37][38][39][40]
The gene was first cloned by scientists at Myriad Genetics, Endo Recherche, Inc., HSC Research & Development Limited Partnership, and the University of Pennsylvania.[41]
Methods to diagnose the likelihood of a patient with mutations in BRCA1 and BRCA2 getting cancer were covered by patents owned or controlled by Myriad Genetics.[42][43] Myriad's business model of exclusively offering the diagnostic test led from Myriad's beginnings as a startup in 1994 to its being a publicly traded company with 1200 employees and about $500M in annual revenue in 2012;[44] it also led to controversy over high test prices and the unavailability of second opinions from other diagnostic labs, which in turn led to the landmark Association for Molecular Pathology v. Myriad Genetics lawsuit.[45]
Germline mutations and founder effect edit
All germline BRCA2 mutations identified to date have been inherited, suggesting the possibility of a large "founder" effect in which a certain mutation is common to a well-defined population group and can theoretically be traced back to a common ancestor. Given the complexity of mutation screening for BRCA2, these common mutations may simplify the methods required for mutation screening in certain populations. Analysis of mutations that occur with high frequency also permits the study of their clinical expression.[46] A striking example of a founder mutation is found in Iceland, where a single BRCA2 (999del5) mutation accounts for virtually all breast/ovarian cancer families.[47][48] This frame-shift mutation leads to a highly truncated protein product. In a large study examining hundreds of cancer and control individuals, this 999del5 mutation was found in 0.6% of the general population. Of note, while 72% of patients who were found to be carriers had a moderate or strong family history of breast cancer, 28% had little or no family history of the disease. This strongly suggests the presence of modifying genes that affect the phenotypic expression of this mutation, or possibly the interaction of the BRCA2 mutation with environmental factors. Additional examples of founder mutations in BRCA2 are given in the table below.
| Population or subgroup | BRCA2 mutation(s)[46][49] | Reference(s) |
|---|---|---|
| Ashkenazi Jewish | 6174delT | [50] |
| Dutch | 5579insA | [51] |
| Finns | 8555T>G, 999del5, IVS23-2A>G | [52][53] |
| French Canadians | 8765delAG, 3398delAAAAG | [54][55][56] |
| Hungarians | 9326insA | [57] |
| Icelanders | 999del5 | [47][48] |
| Italians | 8765delAG | [58] |
| Northern Irish | 6503delTT | [59] |
| Pakistanis | 3337C>T | [60] |
| Scottish | 6503delTT | [59] |
| Slovenians | IVS16-2A>G | [61] |
| Spanish | 3034delAAAC(codon936), 9254del5 | [62] |
| Swedish | 4486delG | [63] |
Meiosis edit
In the plant Arabidopsis thaliana, loss of the BRCA2 homolog AtBRCA2 causes severe defects in both male meiosis and in the development of the female gametocyte.[64] AtBRCA2 protein is required for proper localization of the synaptonemal complex protein AtZYP1 and the recombinases AtRAD51 and AtDMC1. Furthermore, AtBRCA2 is required for proper meiotic synapsis. Thus AtBRCA2 is likely important for meiotic recombination. It appears that AtBRCA2 acts during meiosis to control the single-strand invasion steps mediated by AtRAD51 and AtDMC1 occurring during meiotic homologous recombinational repair of DNA damages.[64]
Homologs of BRCA2 are also essential for meiosis in the fungus Ustilago maydis,[65] the worm Caenorhabditis elegans,[66][67] and the fruitfly Drosophila melanogaster.[68]
Mice that produce truncated versions of BRCA2 are viable but sterile.[69] BRCA2 mutant rats have a phenotype of growth inhibition and sterility in both sexes.[70] Aspermatogenesis in these mutant rats is due to a failure of homologous chromosome synapsis during meiosis.
BRC repeat sequences edit
DMC1 (DNA meiotic recombinase 1) is a meiosis specific homolog of RAD51 that mediates strand exchange during homologous recombinational repair. DMC1 promotes the formation of DNA strand invasion products (joint molecules) between homologous DNA molecules. Human DMC1 interacts directly with each of a series of repeat sequences in the BRCA2 protein (called BRC repeats) that stimulate joint molecule formation by DMC1.[71] BRC repeats conform to a motif consisting of a sequence of about 35 highly conserved amino acids that are present at least once in all BRCA2-like proteins. The BRCA2 BRC repeats stimulate joint molecule formation by promoting the interaction of single-stranded DNA (ssDNA) with DMC1.[71] The ssDNA complexed with DMC1 can pair with homologous ssDNA from another chromosome during the synopsis stage of meiosis to form a joint molecule, a central step in homologous recombination. Thus the BRC repeat sequences of BRCA2 appear to play a key role in recombinational repair of DNA damages during meiotic recombination.
Overall, it appears that homologous recombination during meiosis functions to repair DNA damages,[citation needed] and that BRCA2 plays a key role in performing this function.
Neurogenesis edit
BRCA2 is required in the mouse for neurogenesis and suppression of medulloblastoma.[72] ‘’BRCA2’’ loss profoundly affects neurogenesis, particularly during embryonic and postnatal neural development. These neurological defects arise from DNA damage.[72]
Epigenetic control edit
Epigenetic alterations in expression of BRCA2 (causing over-expression or under-expression) are very frequent in sporadic cancers (see Table below) while mutations in BRCA2 are rarely found.[73][74][75]
In non-small cell lung cancer, BRCA2 is epigenetically repressed by hypermethylation of the promoter.[76] In this case, promoter hypermethylation is significantly associated with low mRNA expression and low protein expression but not with loss of heterozygosity of the gene.
In sporadic ovarian cancer, an opposite effect is found. BRCA2 promoter and 5'-UTR regions have relatively few or no methylated CpG dinucleotides in the tumor DNA compared with that of non-tumor DNA, and a significant correlation is found between hypomethylation and a >3-fold over-expression of BRCA2.[77] This indicates that hypomethylation of the BRCA2 promoter and 5'-UTR regions leads to over-expression of BRCA2 mRNA.
One report indicated some epigenetic control of BRCA2 expression by the microRNAs miR-146a and miR-148a.[78]
BRCA2 expression in cancer edit
In eukaryotes, BRCA2 protein has an important role in homologous recombinational repair. In mice and humans, BRCA2 primarily mediates orderly assembly of RAD51 on single-stranded (ss) DNA, the form that is active for homologous pairing and strand invasion.[79] BRCA2 also redirects RAD51 from double-stranded DNA and prevents dissociation from ssDNA.[79] In addition, the four paralogs of RAD51, consisting of RAD51B (RAD51L1), RAD51C (RAD51L2), RAD51D (RAD51L3), XRCC2 form a complex called the BCDX2 complex (see Figure: Recombinational repair of DNA). This complex participates in RAD51 recruitment or stabilization at damage sites.[26] The BCDX2 complex appears to act by facilitating the assembly or stability of the RAD51 nucleoprotein filament. RAD51 catalyses strand transfer between a broken sequence and its undamaged homologue to allow re-synthesis of the damaged region (see homologous recombination models).
Some studies of cancers report over-expressed BRCA2 whereas other studies report under-expression of BRCA2. At least two reports found over-expression in some sporadic breast tumors and under-expression in other sporadic breast tumors.[80][81] (see Table).
Many cancers have epigenetic deficiencies in various DNA repair genes (see Frequencies of epimutations in DNA repair genes in cancers). These repair deficiencies likely cause increased unrepaired DNA damages. The over-expression of BRCA2 seen in many cancers may reflect compensatory BRCA2 over-expression and increased homologous recombinational repair to at least partially deal with such excess DNA damages. Egawa et al.[82] suggest that increased expression of BRCA2 can be explained by the genomic instability frequently seen in cancers, which induces BRCA2 mRNA expression due to an increased need for BRCA2 for DNA repair.
Under-expression of BRCA2 would itself lead to increased unrepaired DNA damages. Replication errors past these damages (see translesion synthesis) would lead to increased mutations and cancer.
| Cancer | Over or Under expression | Frequency of altered expression | Evaluation method | Ref. |
|---|---|---|---|---|
| Sporadic ovarian cancer | Over-expression | 80% | messenger RNA | [77] |
| Sporadic ovarian cancer | Under-expression | 42% | immunohistochemistry | [83] |
| (recurrent cancer in study above) | Increased-expression | 71% | immunohistochemistry | [83] |
| Non-small cell lung cancer | Under-expression | 34% | immunohistochemistry | [76] |
| Breast cancer | Over-expression | 66% | messenger RNA | [82] |
| Breast cancer | Over-expression | 20% | messenger RNA | [80] |
| (same study as above) | Under-expression | 11% | messenger RNA | [80] |
| Breast cancer | Over-expression | 30% | immunohistochemistry | [81] |
| (same study as above) | Under-expression | 30% | immunohistochemistry | [81] |
| Triple negative breast cancer | Under-expression | 90% | immunohistochemistry | [84] |
Interactions edit
BRCA2 has been shown to interact with
- BRE,[85]
- BARD1,[85][86]
- BCCIP,[87]
- BRCA1,[85][88][89][90]
- BRCC3,[85]
- BUB1B,[91]
- CREBBP,[92]
- C11orf30,[93]
- FANCD2,[94][95][96]
- FANCG,[97]
- FLNA,[98]
- HMG20B,[99][100]
- P53,[85][101]
- PALB2,[29][102]
- PCAF,[103][104]
- PLK1,[103][105]
- RAD51,[85][88][103][106][107][108][109][110][111][112][87][89][101]
- RPA1,[113]
- SHFM1[114][115] and
- SMAD3.[116]
Domain architecture edit
| BRCA2 repeat | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 crystal structure of a rad51-brca2 brc repeat complex | |||||||||
| Identifiers | |||||||||
| Symbol | BRCA2 | ||||||||
| Pfam | PF00634 | ||||||||
| InterPro | IPR002093 | ||||||||
| SCOP2 | 1n0w / SCOPe / SUPFAM | ||||||||
| |||||||||
BRCA2 contains a number of 39 amino acid repeats that are critical for binding to RAD51 (a key protein in DNA recombinational repair) and resistance to methyl methanesulphonate treatment.[101][108][109][117]
The BRCA2 helical domain adopts a helical structure, consisting of a four-helix cluster core (alpha 1, alpha 8, alpha 9, alpha 10) and two successive beta-hairpins (beta 1 to beta 4). An approximately 50-amino acid segment that contains four short helices (alpha 2 to alpha 4), meanders around the surface of the core structure. In BRCA2, the alpha 9 and alpha 10 helices pack with the BRCA2 OB1 domain through van der Waals contacts involving hydrophobic and aromatic residues, and also through side-chain and backbone hydrogen bonds. This domain binds the 70-amino acid DSS1 (deleted in split-hand/split foot syndrome) protein, which was originally identified as one of three genes that map to a 1.5-Mb locus deleted in an inherited developmental malformation syndrome.[115]
The BRCA OB1 domain assumes an OB fold, which consists of a highly curved five-stranded beta-sheet that closes on itself to form a beta-barrel. OB1 has a shallow groove formed by one face of the curved sheet and is demarcated by two loops, one between beta 1 and beta 2 and another between beta 4 and beta 5, which allows for weak single strand DNA binding. The domain also binds the 70-amino acid DSS1 (deleted in split-hand/split foot syndrome) protein.[115]
The BRCA OB3 domain assumes an OB fold, which consists of a highly curved five-stranded beta-sheet that closes on itself to form a beta-barrel. OB3 has a pronounced groove formed by one face of the curved sheet and is demarcated by two loops, one between beta 1 and beta 2 and another between beta 4 and beta 5, which allows for strong ssDNA binding.[115]
The Tower domain adopts a secondary structure consisting of a pair of long, antiparallel alpha-helices (the stem) that support a three-helix bundle (3HB) at their end. The 3HB contains a helix-turn-helix motif and is similar to the DNA binding domains of the bacterial site-specific recombinases, and of eukaryotic Myb and homeodomain transcription factors. The Tower domain has an important role in the tumour suppressor function of BRCA2, and is essential for appropriate binding of BRCA2 to DNA.[115] Studies shown that conformation of this tower domain is allosterically controlled by a small protein "DSS1", which interacts with helical, OB1 and OB2 domains of BRCA2.[118]
Patents, enforcement, litigation, and controversy edit
A patent application for the isolated BRCA1 gene and cancer-cancer promoting mutations, as well as methods to diagnose the likelihood of getting breast cancer, was filed by the University of Utah, National Institute of Environmental Health Sciences (NIEHS) and Myriad Genetics in 1994;[42] over the next year, Myriad, in collaboration with other investigators, isolated and sequenced the BRCA2 gene and identified relevant mutations, and the first BRCA2 patent was filed in the U.S. by Myriad and the other institutions in 1995.[41] Myriad is the exclusive licensee of these patents and has enforced them in the US against clinical diagnostic labs.[45] This business model led from Myriad being a startup in 1994 to being a publicly traded company with 1200 employees and about $500M in annual revenue in 2012;[44] it also led to controversy over high prices and the inability to get second opinions from other diagnostic labs, which in turn led to the landmark Association for Molecular Pathology v. Myriad Genetics lawsuit.[45][119] The patents begin to expire in 2014.
Peter Meldrum, CEO of Myriad Genetics, has acknowledged that Myriad has "other competitive advantages that may make such [patent] enforcement unnecessary" in Europe.[120]
Legal decisions surrounding the BRCA1 and BRCA2 patents will affect the field of genetic testing in general.[121] In June 2013, in Association for Molecular Pathology v. Myriad Genetics (No. 12-398), the US Supreme Court unanimously ruled that, "A naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated," invalidating Myriad's patents on the BRCA1 and BRCA2 genes. However, the Court also held that manipulation of a gene to create something not found in nature could still be eligible for patent protection.[122] The Federal Court of Australia came to the opposite conclusion, upholding the validity of an Australian Myriad Genetics patent over the BRCA1 gene in February 2013,[123] but this decision is being appealed and the appeal will include consideration of the US Supreme Court ruling.[124]
References edit
- ^ a b c GRCh38: Ensembl release 89: ENSG00000139618 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000041147 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Hamel PJ (2007-05-29). "BRCA1 and BRCA2: No Longer the Only Troublesome Genes Out There". HealthCentral. Retrieved 2010-07-02.
- ^ "OrthoMaM phylogenetic marker: BRCA2 coding sequence". Archived from the original on 2016-03-03. Retrieved 2010-02-19.
- ^ "BRCA2 gene tree". Ensembl. May 2021
- ^ Duncan JA, Reeves JR, Cooke TG (October 1998). "BRCA1 and BRCA2 proteins: roles in health and disease". Molecular Pathology. 51 (5): 237–47. doi:10.1136/mp.51.5.237. PMC 395646. PMID 10193517.
- ^ Yoshida K, Miki Y (November 2004). "Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage". Cancer Science. 95 (11): 866–71. doi:10.1111/j.1349-7006.2004.tb02195.x. PMID 15546503. S2CID 24297965.
- ^ Check W (2006-09-01). "BRCA: What we know now". College of American Pathologists. Retrieved 2010-08-23.
- ^ Friedenson B (August 2007). "The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers". BMC Cancer. 7 (1): 152–162. doi:10.1186/1471-2407-7-152. PMC 1959234. PMID 17683622.
- ^ Friedenson B (2008-06-08). "Breast cancer genes protect against some leukemias and lymphomas" (video). SciVee.
- ^ "Breast and Ovarian Cancer Genetic Screening". Palo Alto Medical Foundation. Archived from the original on 4 October 2008. Retrieved 2008-10-11.
- ^ Friedenson B (2007). "The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers". BMC Cancer. 7 (1): 152. doi:10.1186/1471-2407-7-152. PMC 1959234. PMID 17683622.
- ^ O'Donovan PJ, Livingston DM (April 2010). "BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair". Carcinogenesis. 31 (6): 961–7. doi:10.1093/carcin/bgq069. PMID 20400477.
- ^ a b Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. (September 1994). "Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13". Science. 265 (5181): 2088–90. Bibcode:1994Sci...265.2088W. doi:10.1126/science.8091231. PMID 8091231.
- ^ "BRCA2 breast cancer 2, early onset [Homo sapiens]". EntrezGene. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Breast cancer type 2 susceptibility protein - Homo sapiens (Human)". P51587. UniProt.
- ^ Williams-Jones B (2002). Genetic testing for sale: Implications of commercial brca testing in Canada (Ph.D.). The University of British Columbia.
- ^ D'Andrea AD (2010). "Susceptibility pathways in Fanconi's anemia and breast cancer". N. Engl. J. Med. 362 (20): 1909–19. doi:10.1056/NEJMra0809889. PMC 3069698. PMID 20484397.
- ^ Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME (2009). "The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways". J. Biol. Chem. 284 (38): 25560–8. doi:10.1074/jbc.M109.007690. PMC 2757957. PMID 19633289.
- ^ Castillo P, Bogliolo M, Surralles J (2011). "Coordinated action of the Fanconi anemia and ataxia telangiectasia pathways in response to oxidative damage". DNA Repair (Amst.). 10 (5): 518–25. doi:10.1016/j.dnarep.2011.02.007. PMID 21466974.
- ^ Stolz A, Ertych N, Bastians H (2011). "Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability". Clin. Cancer Res. 17 (3): 401–5. doi:10.1158/1078-0432.CCR-10-1215. PMID 21088254.
- ^ Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD (2002). "S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51". Blood. 100 (7): 2414–20. doi:10.1182/blood-2002-01-0278. PMID 12239151.
- ^ Park JY, Zhang F, Andreassen PR (2014). "PALB2: the hub of a network of tumor suppressors involved in DNA damage responses". Biochim. Biophys. Acta. 1846 (1): 263–75. doi:10.1016/j.bbcan.2014.06.003. PMC 4183126. PMID 24998779.
- ^ a b Chun J, Buechelmaier ES, Powell SN (2013). "Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway". Mol. Cell. Biol. 33 (2): 387–95. doi:10.1128/MCB.00465-12. PMC 3554112. PMID 23149936.
- ^ Jensen RB, Carreira A, Kowalczykowski SC (October 2010). "Purified human BRCA2 stimulates RAD51-mediated recombination". Nature. 467 (7316): 678–83. Bibcode:2010Natur.467..678J. doi:10.1038/nature09399. PMC 2952063. PMID 20729832.
- ^ Wang CX, Jimenez-Sainz J, Jensen RB, Mazin AV (March 2019). "The Post-Synaptic Function of Brca2". Scientific Reports. 9 (1): 4554. Bibcode:2019NatSR...9.4554W. doi:10.1038/s41598-019-41054-y. PMC 6418147. PMID 30872704.
- ^ a b Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. (June 2006). "Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2". Molecular Cell. 22 (6): 719–29. doi:10.1016/j.molcel.2006.05.022. PMID 16793542.
- ^ Buisson R, Dion-Côté AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, et al. (October 2010). "Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination". Nature Structural & Molecular Biology. 17 (10): 1247–54. doi:10.1038/nsmb.1915. PMC 4094107. PMID 20871615.
- ^ Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, et al. (October 2017). "Replication fork reversal triggers fork degradation in BRCA2-defective cells". Nature Communications. 8 (1): 859. Bibcode:2017NatCo...8..859M. doi:10.1038/s41467-017-01164-5. PMC 5643541. PMID 29038466.
- ^ Petrucelli N, Daly MB, Pal T (December 2016) [September 1998]. "BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A (eds.). GeneReviews. University of Washington, Seattle. PMID 20301425.
- ^ Levin B, Lech D, Friedenson B (2012). "Evidence that BRCA1- or BRCA2-associated cancers are not inevitable". Molecular Medicine. 18 (9): 1327–37. doi:10.2119/molmed.2012.00280. PMC 3521784. PMID 22972572.
- ^ "High-Penetrance Breast and/or Ovarian Cancer Susceptibility Genes". National Cancer Institute. Retrieved 7 December 2012.
- ^ Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. (1995). "Identification of the breast cancer susceptibility gene BRCA2". Nature. 378 (6559): 789–792. Bibcode:1995Natur.378..789W. doi:10.1038/378789a0. PMID 8524414. S2CID 4346791.
- ^ High-Impact Science: Tracking down the BRCA genes (Part 2) Archived 2012-03-03 at the Wayback Machine - Cancer Research UK science blog, 2012
- ^ "Kenneth Offit | Breast Cancer Research Foundation | BCRF". Bcrfcure.org. 23 June 2014. Retrieved 2015-07-16.
- ^ "A revolution at 50; kenneth offit". The New York Times. 2003-02-25. ISSN 0362-4331. Retrieved 2015-07-02.
- ^ "20 Years of Progress in Understanding Breast Cancer" (JPG). Mskcc.org. Retrieved 2015-07-17.
- ^ Kolata G (1996-10-02). "2d Breast Cancer Gene Found in Jewish Women". The New York Times. ISSN 0362-4331. Retrieved 2015-07-07.
- ^ a b US patent 5837492, Tavtigian SV, Kamb A, Simard J, Couch F, Rommens JM, Weber BL, "Chromosome 13-linked breast cancer susceptibility gene", issued 1998-11-17, assigned to Myriad Genetics, Inc., Endo Recherche, Inc., HSC Research & Development Limited Partnership, Trustees of the University of Pennsylvania
- ^ a b US patent 5747282, Skolnick HS, Goldgar DE, Miki Y, Swenson J, Kamb A, Harshman KD, Shattuck-Eidens DM, Tavtigian SV, Wiseman RW, Futreal PA, "7Q-linked breast and ovarian cancer susceptibility gene", issued 1998-05-05, assigned to Myriad Genetics, Inc., The United States of America as represented by the Secretary of Health and Human Services, and University of Utah Research Foundation
- ^ US patent 5837492, Tavtigian SV, Kamb A, Simard J, Couch F, Rommens JM, Weber BL, "Chromosome 13-linked breast cancer susceptibility gene", issued 1998-11-17, assigned to Myriad Genetics, Inc., Endo Recherche, Inc., HSC Research & Development Limited Partnership, Trustees of the University of Pennsylvania
- ^ a b Myriad Investor Page—see "Myriad at a glance" Archived 2012-10-18 at the Wayback Machine accessed October 2012
- ^ a b c Schwartz J (2009-05-12). "Cancer Patients Challenge the Patenting of a Gene". Health. New York Times.
- ^ a b Lacroix M, Leclercq G (2005). "The "portrait" of hereditary breast cancer". Breast Cancer Research and Treatment. 89 (3): 297–304. doi:10.1007/s10549-004-2172-4. PMID 15754129. S2CID 23327569.
- ^ a b Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson JG, Tavtigian SV, et al. (1996). "A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes". Nature Genetics. 13 (1): 117–119. doi:10.1038/ng0596-117. PMID 8673089. S2CID 8443452.
- ^ a b Thorlacius S, Sigurdsson S, Bjarnadottir H, Olafsdottir G, Jonasson JG, Tryggvadottir L, et al. (1997). "Study of a single BRCA2 mutation with high carrier frequency in a small population". American Journal of Human Genetics. 60 (5): 1079–1085. PMC 1712443. PMID 9150155.
- ^ den Dunnen JT, Antonarakis SE (2000). "Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion". Human Mutation. 15 (1): 7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. PMID 10612815.
- ^ Neuhausen S, Gilewski T, Norton L, Tran T, McGuire P, Swensen J, et al. (1996). "Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer". Nature Genetics. 13 (1): 126–128. doi:10.1038/ng0596-126. PMID 8673092. S2CID 11909356.
- ^ Verhoog LC, van den Ouweland AM, Berns E, van Veghel-Plandsoen MM, van Staveren IL, Wagner A, et al. (2001). "Large regional differences in the frequency of distinct BRCA1/BRCA2 mutations in 517 Dutch breast and/or ovarian cancer families". European Journal of Cancer. 37 (16): 2082–2090. doi:10.1016/S0959-8049(01)00244-1. PMID 11597388.
- ^ Huusko P, Pääkkönen K, Launonen V, Pöyhönen M, Blanco G, Kauppila A, et al. (1998). "Evidence of founder mutations in Finnish BRCA1 and BRCA2 families". American Journal of Human Genetics. 62 (6): 1544–1548. doi:10.1086/301880. PMC 1377159. PMID 9585608.
- ^ Pääkkönen K, Sauramo S, Sarantaus L, Vahteristo P, Hartikainen A, Vehmanen P, et al. (2001). "Involvement of BRCA1 and BRCA2 in breast cancer in a western Finnish sub-population". Genetic Epidemiology. 20 (2): 239–246. doi:10.1002/1098-2272(200102)20:2<239::AID-GEPI6>3.0.CO;2-Y. PMID 11180449. S2CID 41804152.
- ^ Tonin PN, Mes-Masson AM, Narod SA, Ghadirian P, Provencher D (1999). "Founder BRCA1 and BRCA2 mutations in French Canadian ovarian cancer cases unselected for family history". Clinical Genetics. 55 (5): 318–324. doi:10.1034/j.1399-0004.1999.550504.x. PMID 10422801. S2CID 23931343.
- ^ Oros KK, Leblanc G, Arcand SL, Shen Z, Perret C, Mes-Masson AM, et al. (2006). "Haplotype analysis suggests common founders in carriers of recurrent BRCA2 mutation, 3398delAAAAG, in French Canadian hereditary breast and/ovarian cancer families". BMC Medical Genetics. 7 (23): 23. doi:10.1186/1471-2350-7-23. PMC 1464093. PMID 16539696.
- ^ Tonin PN (2006). "The limited spectrum of pathogenic BRCA1 and BRCA2 mutations in the French Canadian breast and breast-ovarian cancer families, a founder population of Quebec, Canada". Bull Cancer. 93 (9): 841–846. PMID 16980226.
- ^ Van Der Looij M, Szabo C, Besznyak I, Liszka G, Csokay B, Pulay T, et al. (2000). "Prevalence of founder BRCA1 and BRCA2 mutations among breast and ovarian cancer patients in Hungary". International Journal of Cancer. 86 (5): 737–740. doi:10.1002/(SICI)1097-0215(20000601)86:5<737::AID-IJC21>3.0.CO;2-1. PMID 10797299. S2CID 25394976.
- ^ Pisano M, Cossu A, Persico I, Palmieri G, Angius A, Casu G, et al. (2000). "Identification of a founder BRCA2 mutation in Sardinia". British Journal of Cancer. 82 (3): 553–559. doi:10.1054/bjoc.1999.0963. PMC 2363305. PMID 10682665.
- ^ a b Scottish/Northern Irish BRCAI/BRCA2 Consortium (2003). "BRCA1 and BRCA2 mutations in Scotland and Northern Ireland". British Journal of Cancer. 88 (8): 1256–1262. doi:10.1038/sj.bjc.6600840. PMC 2747571. PMID 12698193.
{{cite journal}}: CS1 maint: numeric names: authors list (link) - ^ Liede A, Malik IA, Aziz Z, Rios Pd Pde L, Kwan E, Narod SA (2002). "Contribution of BRCA1 and BRCA2 mutations to breast and ovarian cancer in Pakistan". American Journal of Human Genetics. 71 (3): 595–606. doi:10.1086/342506. PMC 379195. PMID 12181777.
- ^ Krajc M, De Grève J, Goelen G, Teugels E (2002). "BRCA2 founder mutation in Slovenian breast cancer families". European Journal of Human Genetics. 10 (12): 879–882. doi:10.1038/sj.ejhg.5200886. PMID 12461697.
- ^ Osorio A, Robledo M, Martínez B, Cebrián A, San Román JM, Albertos J, et al. (1998). "Molecular analysis of the BRCA2 gene in 16 breast/ovarian cancer Spanish families". Clin. Genet. 54 (2): 142–7. doi:10.1111/j.1399-0004.1998.tb03717.x. PMID 9761393. S2CID 30388365.
- ^ Neuhausen SL (2000). "Founder populations and their uses for breast cancer genetics". Cancer Research. 2 (2): 77–81. doi:10.1186/bcr36. PMC 139426. PMID 11250694.
- ^ a b Seeliger K, Dukowic-Schulze S, Wurz-Wildersinn R, Pacher M, Puchta H (2012). "BRCA2 is a mediator of RAD51- and DMC1-facilitated homologous recombination in Arabidopsis thaliana". New Phytol. 193 (2): 364–75. doi:10.1111/j.1469-8137.2011.03947.x. PMID 22077663.
- ^ Kojic M, Kostrub CF, Buchman AR, Holloman WK (2002). "BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis". Mol. Cell. 10 (3): 683–91. doi:10.1016/s1097-2765(02)00632-9. PMID 12408834.
- ^ Ko E, Lee J, Lee H (2008). "Essential role of brc-2 in chromosome integrity of germ cells in C. elegans". Mol. Cells. 26 (6): 590–4. doi:10.1016/S1016-8478(23)14041-6. PMID 18779660.
- ^ Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, Boulton SJ (2005). "RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair". Mol. Cell. Biol. 25 (8): 3127–39. doi:10.1128/MCB.25.8.3127-3139.2005. PMC 1069622. PMID 15798199.
- ^ Klovstad M, Abdu U, Schüpbach T (2008). "Drosophila brca2 is required for mitotic and meiotic DNA repair and efficient activation of the meiotic recombination checkpoint". PLOS Genet. 4 (2): e31. doi:10.1371/journal.pgen.0040031. PMC 2233675. PMID 18266476.
- ^ Connor F, Bertwistle D, Mee PJ, Ross GM, Swift S, Grigorieva E, et al. (1997). "Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation". Nat. Genet. 17 (4): 423–30. doi:10.1038/ng1297-423. PMID 9398843. S2CID 42462448.
- ^ Cotroneo MS, Haag JD, Zan Y, Lopez CC, Thuwajit P, Petukhova GV, et al. (2007). "Characterizing a rat Brca2 knockout model". Oncogene. 26 (11): 1626–35. doi:10.1038/sj.onc.1209960. PMID 16964288.
- ^ a b Martinez JS, von Nicolai C, Kim T, Ehlén Å, Mazin AV, Kowalczykowski SC, et al. (2016). "BRCA2 regulates DMC1-mediated recombination through the BRC repeats". Proc. Natl. Acad. Sci. U.S.A. 113 (13): 3515–20. Bibcode:2016PNAS..113.3515M. doi:10.1073/pnas.1601691113. PMC 4822569. PMID 26976601.
- ^ a b Frappart PO, Lee Y, Lamont J, McKinnon PJ (2007). "BRCA2 is required for neurogenesis and suppression of medulloblastoma". EMBO J. 26 (11): 2732–42. doi:10.1038/sj.emboj.7601703. PMC 1888666. PMID 17476307.
- ^ Teng DH, Bogden R, Mitchell J, Baumgard M, Bell R, Berry S, et al. (1996). "Low incidence of BRCA2 mutations in breast carcinoma and other cancers". Nat. Genet. 13 (2): 241–4. doi:10.1038/ng0696-241. PMID 8640236. S2CID 9831745.
- ^ Miki Y, Katagiri T, Kasumi F, Yoshimoto T, Nakamura Y (1996). "Mutation analysis in the BRCA2 gene in primary breast cancers". Nat. Genet. 13 (2): 245–7. doi:10.1038/ng0696-245. PMID 8640237. S2CID 3203046.
- ^ Lancaster JM, Wooster R, Mangion J, Phelan CM, Cochran C, Gumbs C, et al. (1996). "BRCA2 mutations in primary breast and ovarian cancers". Nat. Genet. 13 (2): 238–40. doi:10.1038/ng0696-238. PMID 8640235. S2CID 26808443.
- ^ a b Lee MN, Tseng RC, Hsu HS, Chen JY, Tzao C, Ho WL, et al. (2007). "Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer". Clin. Cancer Res. 13 (3): 832–8. doi:10.1158/1078-0432.CCR-05-2694. PMID 17289874.
- ^ a b Chan KY, Ozçelik H, Cheung AN, Ngan HY, Khoo US (2002). "Epigenetic factors controlling the BRCA1 and BRCA2 genes in sporadic ovarian cancer". Cancer Res. 62 (14): 4151–6. PMID 12124354.
- ^ Gu Y, Zhang M, Peng F, Fang L, Zhang Y, Liang H, et al. (2015). "The BRCA1/2-directed miRNA signature predicts a good prognosis in ovarian cancer patients with wild-type BRCA1/2". Oncotarget. 6 (4): 2397–406. doi:10.18632/oncotarget.2963. PMC 4385859. PMID 25537514.
- ^ a b Holloman WK (2011). "Unraveling the mechanism of BRCA2 in homologous recombination". Nat. Struct. Mol. Biol. 18 (7): 748–54. doi:10.1038/nsmb.2096. PMC 3647347. PMID 21731065.
- ^ a b c Bièche I, Noguès C, Lidereau R (1999). "Overexpression of BRCA2 gene in sporadic breast tumours". Oncogene. 18 (37): 5232–8. doi:10.1038/sj.onc.1202903. PMID 10498873.
- ^ a b c Hedau S, Batra M, Singh UR, Bharti AC, Ray A, Das BC (2015). "Expression of BRCA1 and BRCA2 proteins and their correlation with clinical staging in breast cancer". J Cancer Res Ther. 11 (1): 158–63. doi:10.4103/0973-1482.140985. PMID 25879355.
- ^ a b Egawa C, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S (2002). "High BRCA2 mRNA expression predicts poor prognosis in breast cancer patients". Int. J. Cancer. 98 (6): 879–82. doi:10.1002/ijc.10231. PMID 11948466. S2CID 9083282.
- ^ a b Swisher EM, Gonzalez RM, Taniguchi T, Garcia RL, Walsh T, Goff BA, et al. (2009). "Methylation and protein expression of DNA repair genes: association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas". Mol. Cancer. 8 (1): 48. doi:10.1186/1476-4598-8-48. PMC 2719582. PMID 19602291.
- ^ Thike AA, Tan PH, Ikeda M, Iqbal J (2016). "Increased ID4 expression, accompanied by mutant p53 accumulation and loss of BRCA1/2 proteins in triple-negative breast cancer, adversely affects survival". Histopathology. 68 (5): 702–12. doi:10.1111/his.12801. PMID 26259780. S2CID 3566545.
- ^ a b c d e f Dong Y, Hakimi MA, Chen X, Kumaraswamy E, Cooch NS, Godwin AK, et al. (November 2003). "Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair". Mol. Cell. 12 (5): 1087–99. doi:10.1016/S1097-2765(03)00424-6. PMID 14636569.
- ^ Ryser S, Dizin E, Jefford CE, Delaval B, Gagos S, Christodoulidou A, et al. (February 2009). "Distinct roles of BARD1 isoforms in mitosis: full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2". Cancer Res. 69 (3): 1125–34. doi:10.1158/0008-5472.CAN-08-2134. PMID 19176389.
- ^ a b Liu J, Yuan Y, Huan J, Shen Z (January 2001). "Inhibition of breast and brain cancer cell growth by BCCIPalpha, an evolutionarily conserved nuclear protein that interacts with BRCA2". Oncogene. 20 (3): 336–45. doi:10.1038/sj.onc.1204098. PMID 11313963.
- ^ a b Sarkisian CJ, Master SR, Huber LJ, Ha SI, Chodosh LA (October 2001). "Analysis of murine Brca2 reveals conservation of protein-protein interactions but differences in nuclear localization signals". J. Biol. Chem. 276 (40): 37640–8. doi:10.1074/jbc.M106281200. PMID 11477095.
- ^ a b Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, et al. (September 1998). "Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells". Mol. Cell. 2 (3): 317–28. doi:10.1016/S1097-2765(00)80276-2. PMID 9774970.
- ^ Reuter TY, Medhurst AL, Waisfisz Q, Zhi Y, Herterich S, Hoehn H, et al. (October 2003). "Yeast two-hybrid screens imply involvement of Fanconi anemia proteins in transcription regulation, cell signaling, oxidative metabolism, and cellular transport". Exp. Cell Res. 289 (2): 211–21. doi:10.1016/S0014-4827(03)00261-1. PMID 14499622.
- ^ Futamura M, Arakawa H, Matsuda K, Katagiri T, Saji S, Miki Y, et al. (March 2000). "Potential role of BRCA2 in a mitotic checkpoint after phosphorylation by hBUBR1". Cancer Res. 60 (6): 1531–5. PMID 10749118.
- ^ Siddique H, Rao VN, Reddy ES (August 2009). "CBP-mediated post-translational N-glycosylation of BRCA2". Int J Oncol. 35 (2): 16387–91. doi:10.3892/ijo_00000351. PMID 19578754.
- ^ Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin SF, et al. (November 2003). "EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer". Cell. 115 (5): 523–35. doi:10.1016/S0092-8674(03)00930-9. PMID 14651845. S2CID 18911371.
- ^ Wang X, Andreassen PR, D'Andrea AD (July 2004). "Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin". Mol. Cell. Biol. 24 (13): 5850–62. doi:10.1128/MCB.24.13.5850-5862.2004. PMC 480901. PMID 15199141.
- ^ Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, et al. (June 2004). "Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways". Hum. Mol. Genet. 13 (12): 1241–8. doi:10.1093/hmg/ddh135. PMID 15115758.
- ^ Hejna J, Holtorf M, Hines J, Mathewson L, Hemphill A, Al-Dhalimy M, et al. (April 2008). "Tip60 is required for DNA interstrand cross-link repair in the Fanconi anemia pathway". J. Biol. Chem. 283 (15): 9844–51. doi:10.1074/jbc.M709076200. PMC 2398728. PMID 18263878.
- ^ Hussain S, Witt E, Huber PA, Medhurst AL, Ashworth A, Mathew CG (October 2003). "Direct interaction of the Fanconi anaemia protein FANCG with BRCA2/FANCD1". Hum. Mol. Genet. 12 (19): 2503–10. doi:10.1093/hmg/ddg266. PMID 12915460.
- ^ Yuan Y, Shen Z (December 2001). "Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response". J. Biol. Chem. 276 (51): 48318–24. doi:10.1074/jbc.M102557200. PMID 11602572.
- ^ Marmorstein LY, Kinev AV, Chan GK, Bochar DA, Beniya H, Epstein JA, et al. (January 2001). "A human BRCA2 complex containing a structural DNA binding component influences cell cycle progression". Cell. 104 (2): 247–57. doi:10.1016/S0092-8674(01)00209-4. PMID 11207365. S2CID 5822368.
- ^ Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R (May 2002). "A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes". Proc. Natl. Acad. Sci. U.S.A. 99 (11): 7420–5. Bibcode:2002PNAS...99.7420H. doi:10.1073/pnas.112008599. PMC 124246. PMID 12032298.
- ^ a b c Marmorstein LY, Ouchi T, Aaronson SA (November 1998). "The BRCA2 gene product functionally interacts with p53 and RAD51". Proc. Natl. Acad. Sci. U.S.A. 95 (23): 13869–74. Bibcode:1998PNAS...9513869M. doi:10.1073/pnas.95.23.13869. PMC 24938. PMID 9811893.
- ^ "Entrez Gene: PALB2 partner and localizer of BRCA2".
- ^ a b c Lin HR, Ting NS, Qin J, Lee WH (September 2003). "M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex". J. Biol. Chem. 278 (38): 35979–87. doi:10.1074/jbc.M210659200. PMID 12815053.
- ^ Fuks F, Milner J, Kouzarides T (November 1998). "BRCA2 associates with acetyltransferase activity when bound to P/CAF". Oncogene. 17 (19): 2531–4. doi:10.1038/sj.onc.1202475. PMID 9824164.
- ^ Lee M, Daniels MJ, Venkitaraman AR (January 2004). "Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression". Oncogene. 23 (4): 865–72. doi:10.1038/sj.onc.1207223. PMID 14647413.
- ^ Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, et al. (April 1997). "Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2". Nature. 386 (6627): 804–10. Bibcode:1997Natur.386..804S. doi:10.1038/386804a0. hdl:11858/00-001M-0000-0010-5059-F. PMID 9126738. S2CID 4238943.
- ^ Yu DS, Sonoda E, Takeda S, Huang CL, Pellegrini L, Blundell TL, et al. (October 2003). "Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2". Mol. Cell. 12 (4): 1029–41. doi:10.1016/S1097-2765(03)00394-0. PMID 14580352.
- ^ a b Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH (April 1998). "The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment". Proc. Natl. Acad. Sci. U.S.A. 95 (9): 5287–92. Bibcode:1998PNAS...95.5287C. doi:10.1073/pnas.95.9.5287. PMC 20253. PMID 9560268.
- ^ a b Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL (December 1997). "RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2". J. Biol. Chem. 272 (51): 31941–4. doi:10.1074/jbc.272.51.31941. PMID 9405383.
- ^ Katagiri T, Saito H, Shinohara A, Ogawa H, Kamada N, Nakamura Y, et al. (March 1998). "Multiple possible sites of BRCA2 interacting with DNA repair protein RAD51". Genes Chromosomes Cancer. 21 (3): 217–22. doi:10.1002/(SICI)1098-2264(199803)21:3<217::AID-GCC5>3.0.CO;2-2. PMID 9523196. S2CID 45954246.
- ^ Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, et al. (November 2002). "Insights into DNA recombination from the structure of a RAD51-BRCA2 complex". Nature. 420 (6913): 287–93. Bibcode:2002Natur.420..287P. doi:10.1038/nature01230. PMID 12442171. S2CID 4359383.
- ^ Tarsounas M, Davies AA, West SC (January 2004). "RAD51 localization and activation following DNA damage". Philos. Trans. R. Soc. Lond. B Biol. Sci. 359 (1441): 87–93. doi:10.1098/rstb.2003.1368. PMC 1693300. PMID 15065660.
- ^ Wong JM, Ionescu D, Ingles CJ (January 2003). "Interaction between BRCA2 and replication protein A is compromised by a cancer-predisposing mutation in BRCA2". Oncogene. 22 (1): 28–33. doi:10.1038/sj.onc.1206071. PMID 12527904.
- ^ Marston NJ, Richards WJ, Hughes D, Bertwistle D, Marshall CJ, Ashworth A (July 1999). "Interaction between the product of the breast cancer susceptibility gene BRCA2 and DSS1, a protein functionally conserved from yeast to mammals". Mol. Cell. Biol. 19 (7): 4633–42. doi:10.1128/MCB.19.7.4633. PMC 84261. PMID 10373512.
- ^ a b c d e Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, et al. (September 2002). "BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure". Science. 297 (5588): 1837–48. Bibcode:2002Sci...297.1837Y. doi:10.1126/science.297.5588.1837. PMID 12228710.
- ^ Preobrazhenska O, Yakymovych M, Kanamoto T, Yakymovych I, Stoika R, Heldin CH, et al. (August 2002). "BRCA2 and Smad3 synergize in regulation of gene transcription". Oncogene. 21 (36): 5660–4. doi:10.1038/sj.onc.1205732. PMID 12165866.
- ^ Bork P, Blomberg N, Nilges M (May 1996). "Internal repeats in the BRCA2 protein sequence". Nat. Genet. 13 (1): 22–3. doi:10.1038/ng0596-22. PMID 8673099. S2CID 2312211.
- ^ Alagar S, Bahadur RP (2020). "DSS1 allosterically regulates the conformation of the tower domain of BRCA2 that has dsDNA binding specificity for homologous recombination". International Journal of Biological Macromolecules. 165 (Pt A): 918–929. doi:10.1016/j.ijbiomac.2020.09.230. PMID 33011260. S2CID 222165754.
- ^ "ACLU sues over patents on breast cancer genes". CNN. Archived from the original on 15 May 2009. Retrieved 2009-05-14.
- ^ Conley J, Vorhous D, Cook-Deegan J (2011-03-01). "How Will Myriad Respond to the Next Generation of BRCA Testing?". Robinson, Bradshaw, and Hinson. Retrieved 2012-12-09.
- ^ "Genetics and Patenting". Human Genome Project Information. U.S. Department of Energy Genome Programs. 2010-07-07.
- ^ Liptak A (13 June 2013). "Supreme Court Rules Human Genes May Not Be Patented". New York Times. Retrieved 13 June 2013.
- ^ Corderoy A (February 15, 2013). "Landmark patent ruling over breast cancer gene BRCA1". Sydney Morning Herald. Retrieved June 14, 2013.
- ^ Corderoy A (June 14, 2013). "Companies can't patent genes, US court rules". Sydney Morning Herald. Retrieved June 14, 2013.
{kind=link}
Further reading edit
- Zou JP, Hirose Y, Siddique H, Rao VN, Reddy ES (1999). "Structure and expression of variant BRCA2a lacking the transactivation domain". Oncology Reports. 6 (2): 437–40. doi:10.3892/or.6.2.437. PMID 10023017.
- Venkitaraman AR (2001). "Chromosome stability, DNA recombination and the BRCA2 tumour suppressor". Current Opinion in Cell Biology. 13 (3): 338–43. doi:10.1016/S0955-0674(00)00217-9. PMID 11343905.
- Orelli BJ, Bishop DK (2001). "BRCA2 and homologous recombination". Breast Cancer Research. 3 (5): 294–8. doi:10.1186/bcr310. PMC 138691. PMID 11597317.
- Daniel DC (2002). "Highlight: BRCA1 and BRCA2 proteins in breast cancer". Microscopy Research and Technique. 59 (1): 68–83. doi:10.1002/jemt.10178. PMID 12242698. S2CID 30091586.
- Tutt A, Ashworth A (2003). "The relationship between the roles of BRCA genes in DNA repair and cancer predisposition". Trends in Molecular Medicine. 8 (12): 571–6. doi:10.1016/S1471-4914(02)02434-6. PMID 12470990.
- Gonçalves A, Viens P, Sobol H, Maraninchi D, Bertucci F (2005). "[Molecular alterations in breast cancer: clinical implications and new analytical tools]". Revue de Médecine Interne. 26 (6): 470–8. doi:10.1016/j.revmed.2004.11.012. PMID 15936476.
- Hay T, Clarke AR (2005). "DNA damage hypersensitivity in cells lacking BRCA2: a review of in vitro and in vivo data". Biochemical Society Transactions. 33 (Pt 4): 715–7. doi:10.1042/BST0330715. PMID 16042582.
- Domchek SM, Weber BL (2006). "Clinical management of BRCA1 and BRCA2 mutation carriers". Oncogene. 25 (43): 5825–31. doi:10.1038/sj.onc.1209881. PMID 16998496.
- Honrado E, Osorio A, Palacios J, Benitez J (2006). "Pathology and gene expression of hereditary breast tumors associated with BRCA1, BRCA2 and CHEK2 gene mutations". Oncogene. 25 (43): 5837–45. doi:10.1038/sj.onc.1209875. PMID 16998498. S2CID 20960561.
External links edit
- BRCA2 Protein at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
|}