Cytochromes P450 (P450s or CYPs) are a superfamily of enzymes containing heme as a cofactor that mostly, but not exclusively, function as monooxygenases.[1][2][3] In mammals, these proteins oxidize steroids, fatty acids, and xenobiotics, and are important for the clearance of various compounds, as well as for hormone synthesis and breakdown, steroid hormone synthesis, drug metabolism, and the biosynthesis of defensive compounds, fatty acids, and hormones.[2] CYP450 enzymes convert xenobiotics into hydrophilic derivatives, which are more readily excreted. In almost all of the transformations that they catalyze, P450's affect hydroxylation.
| Cytochrome P450 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
.png) Structure of lanosterol 14α-demethylase (CYP51) | |||||||||
| Identifiers | |||||||||
| Symbol | p450 | ||||||||
| Pfam | PF00067 | ||||||||
| InterPro | IPR001128 | ||||||||
| PROSITE | PDOC00081 | ||||||||
| SCOP2 | 2cpp / SCOPe / SUPFAM | ||||||||
| OPM superfamily | 39 | ||||||||
| OPM protein | 2bdm | ||||||||
| CDD | cd00302 | ||||||||
| Membranome | 265 | ||||||||
| |||||||||
P450 enzymes have been identified in all kingdoms of life: animals, plants, fungi, protists, bacteria, and archaea, as well as in viruses.[4] However, they are not omnipresent; for example, they have not been found in Escherichia coli.[3][5] As of 2018[update], more than 300,000 distinct CYP proteins are known.[6][7]
P450s are, in general, the terminal oxidase enzymes in electron transfer chains, broadly categorized as P450-containing systems. The term "P450" is derived from the spectrophotometric peak at the wavelength of the absorption maximum of the enzyme (450 nm) when it is in the reduced state and complexed with carbon monoxide. Most P450s require a protein partner to deliver one or more electrons to reduce the iron (and eventually molecular oxygen).
Nomenclature edit
Genes encoding P450 enzymes, and the enzymes themselves, are designated with the root symbol CYP for the superfamily, followed by a number indicating the gene family, a capital letter indicating the subfamily, and another numeral for the individual gene. The convention is to italicise the name when referring to the gene. For example, CYP2E1 is the gene that encodes the enzyme CYP2E1—one of the enzymes involved in paracetamol (acetaminophen) metabolism. The CYP nomenclature is the official naming convention, although occasionally CYP450 or CYP450 is used synonymously. These names should never be used as according to the nomenclature convention (as they denote a P450 in family number 450). However, some gene or enzyme names for P450s are also referred to by historical names (e.g. P450BM3 for CYP102A1) or functional names, denoting the catalytic activity and the name of the compound used as substrate. Examples include CYP5A1, thromboxane A2 synthase, abbreviated to TBXAS1 (ThromBoXane A2 Synthase 1), and CYP51A1, lanosterol 14-α-demethylase, sometimes unofficially abbreviated to LDM according to its substrate (Lanosterol) and activity (DeMethylation).[8]
The current nomenclature guidelines suggest that members of new CYP families share at least 40% amino-acid identity, while members of subfamilies must share at least 55% amino-acid identity. Nomenclature committees assign and track both base gene names (Cytochrome P450 Homepage Archived 2010-06-27 at the Wayback Machine) and allele names (CYP Allele Nomenclature Committee).[9][10]
Classification edit
Based on the nature of the electron transfer proteins, P450s can be classified into several groups:[11]
- Microsomal P450 systems
- in which electrons are transferred from NADPH via cytochrome P450 reductase (variously CPR, POR, or CYPOR). Cytochrome b5 (cyb5) can also contribute reducing power to this system after being reduced by cytochrome b5 reductase (CYB5R).
- Mitochondrial P450 systems
- which employ adrenodoxin reductase and adrenodoxin to transfer electrons from NADPH to P450.
- Bacterial P450 systems
- which employ a ferredoxin reductase and a ferredoxin to transfer electrons to P450.
- CYB5R/cyb5/P450 systems
- in which both electrons required by the CYP come from cytochrome b5.
- FMN/Fd/P450 systems
- originally found in Rhodococcus species, in which a FMN-domain-containing reductase is fused to the CYP.
- P450 only systems
- which do not require external reducing power. Notable ones include thromboxane synthase (CYP5), prostacyclin synthase (CYP8), and CYP74A (allene oxide synthase).
The most common reaction catalyzed by cytochromes P450 is a monooxygenase reaction, e.g., insertion of one atom of oxygen into the aliphatic position of an organic substrate (RH), while the other oxygen atom is reduced to water:
Many hydroxylation reactions (insertion of hydroxyl groups) use CYP enzymes.
Mechanism edit

Structure edit
The active site of cytochrome P450 contains a heme-iron center. The iron is tethered to the protein via a cysteine thiolate ligand. This cysteine and several flanking residues are highly conserved in known P450s, and have the formal PROSITE signature consensus pattern [FW] - [SGNH] - x - [GD] - {F} - [RKHPT] - {P} - C - [LIVMFAP] - [GAD].[12] Because of the vast variety of reactions catalyzed by P450s, the activities and properties of the many P450s differ in many aspects. In general, the P450 catalytic cycle proceeds as follows:
Catalytic cycle edit
- Substrate binds in proximity to the heme group, on the side opposite to the axial thiolate. Substrate binding induces a change in the conformation of the active site, often displacing a water molecule from the distal axial coordination position of the heme iron,[13] and changing the state of the heme iron from low-spin to high-spin.[14]
- Substrate binding induces electron transfer from NAD(P)H via cytochrome P450 reductase or another associated reductase.[15]
- Molecular oxygen binds to the resulting ferrous heme center at the distal axial coordination position, initially giving a dioxygen adduct similar to oxy-myoglobin.
- A second electron is transferred, from either cytochrome P450 reductase, ferredoxins, or cytochrome b5, reducing the Fe-O2 adduct to give a short-lived peroxo state.
- The peroxo group formed in step 4 is rapidly protonated twice, releasing one molecule of water and forming the highly reactive species referred to as P450 Compound 1 (or just Compound I). This highly reactive intermediate was isolated in 2010,[16] P450 Compound 1 is an iron(IV) oxo (or ferryl) species with an additional oxidizing equivalent delocalized over the porphyrin and thiolate ligands. Evidence for the alternative perferryl iron(V)-oxo[13] is lacking.[16]
- Depending on the substrate and enzyme involved, P450 enzymes can catalyze any of a wide variety of reactions. A hypothetical hydroxylation is shown in this illustration. After the product has been released from the active site, the enzyme returns to its original state, with a water molecule returning to occupy the distal coordination position of the iron nucleus.
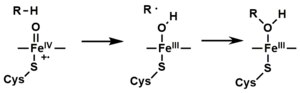
- An alternative route for mono-oxygenation is via the "peroxide shunt" (path "S" in figure). This pathway entails oxidation of the ferric-substrate complex with oxygen-atom donors such as peroxides and hypochlorites.[17] A hypothetical peroxide "XOOH" is shown in the diagram.
Spectroscopy edit
Binding of substrate is reflected in the spectral properties of the enzyme, with an increase in absorbance at 390 nm and a decrease at 420 nm. This can be measured by difference spectroscopies and is referred to as the "type I" difference spectrum (see inset graph in figure). Some substrates cause an opposite change in spectral properties, a "reverse type I" spectrum, by processes that are as yet unclear. Inhibitors and certain substrates that bind directly to the heme iron give rise to the type II difference spectrum, with a maximum at 430 nm and a minimum at 390 nm (see inset graph in figure). If no reducing equivalents are available, this complex may remain stable, allowing the degree of binding to be determined from absorbance measurements in vitro[17] C: If carbon monoxide (CO) binds to reduced P450, the catalytic cycle is interrupted. This reaction yields the classic CO difference spectrum with a maximum at 450 nm. However, the interruptive and inhibitory effects of CO varies upon different CYPs such that the CYP3A family is relatively less affected.[18]
P450s in humans edit
Human P450s are primarily membrane-associated proteins[19] located either in the inner membrane of mitochondria or in the endoplasmic reticulum of cells. P450s metabolize thousands of endogenous and exogenous chemicals. Some P450s metabolize only one (or a very few) substrates, such as CYP19 (aromatase), while others may metabolize multiple substrates. Both of these characteristics account for their central importance in medicine. Cytochrome P450 enzymes are present in most tissues of the body, and play important roles in hormone synthesis and breakdown (including estrogen and testosterone synthesis and metabolism), cholesterol synthesis, and vitamin D metabolism. Cytochrome P450 enzymes also function to metabolize potentially toxic compounds, including drugs and products of endogenous metabolism such as bilirubin, principally in the liver.
The Human Genome Project has identified 57 human genes coding for the various cytochrome P450 enzymes.[20]
Drug metabolism edit

P450s are the major enzymes involved in drug metabolism, accounting for about 75% of the total metabolism.[22] Most drugs undergo deactivation by P450s, either directly or by facilitated excretion from the body. However, many substances are bioactivated by P450s to form their active compounds like the antiplatelet drug clopidogrel and the opiate codeine.
The CYP450 enzyme superfamily comprises 57 active subsets, with seven playing a crucial role in the metabolism of most pharmaceuticals.[23] The fluctuation in the amount of CYP450 enzymes (CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5) in phase 1 (detoxification) can have varying effects on individuals, as genetic expression varies from person to person. This variation is due to the enzyme’s genetic polymorphism, which leads to variability in its function and expression. To optimize drug metabolism in individuals, genetic testing should be conducted to determine functional foods and specific phytonutrients that cater to the individual’s CYP450 polymorphism. Understanding these genetic variations can help personalize drug therapies for improved effectiveness and reduced adverse reactions.[24]
Drug interaction edit
Many drugs may increase or decrease the activity of various P450 isozymes either by inducing the biosynthesis of an isozyme (enzyme induction) or by directly inhibiting the activity of the P450 (enzyme inhibition). A classical example includes anti-epileptic drugs, such as phenytoin, which induces CYP1A2, CYP2C9, CYP2C19, and CYP3A4.
Effects on P450 isozyme activity are a major source of adverse drug interactions, since changes in P450 enzyme activity may affect the metabolism and clearance of various drugs. For example, if one drug inhibits the P450-mediated metabolism of another drug, the second drug may accumulate within the body to toxic levels. Hence, these drug interactions may necessitate dosage adjustments or choosing drugs that do not interact with the P450 system. Such drug interactions are especially important to consider when using drugs of vital importance to the patient, drugs with significant side-effects, or drugs with a narrow therapeutic index, but any drug may be subject to an altered plasma concentration due to altered drug metabolism.
Many substrates for CYP3A4 are drugs with a narrow therapeutic index, such as amiodarone[25] or carbamazepine.[26] Because these drugs are metabolized by CYP3A4, the mean plasma levels of these drugs may increase because of enzyme inhibition or decrease because of enzyme induction.
Interaction of other substances edit
Naturally occurring compounds may also induce or inhibit P450 activity. For example, bioactive compounds found in grapefruit juice and some other fruit juices, including bergamottin, dihydroxybergamottin, and paradicin-A, have been found to inhibit CYP3A4-mediated metabolism of certain medications, leading to increased bioavailability and, thus, the strong possibility of overdosing.[27] Because of this risk, avoiding grapefruit juice and fresh grapefruits entirely while on drugs is usually advised.[28]
Other examples:
- Saint-John's wort, a common herbal remedy induces CYP3A4, but also inhibits CYP1A1, CYP1B1.[29][30]
- Tobacco smoking induces CYP1A2 (example CYP1A2 substrates are clozapine, olanzapine, and fluvoxamine)[31]
- At relatively high concentrations, starfruit juice has also been shown to inhibit CYP2A6 and other P450s.[32] Watercress is also a known inhibitor of the cytochrome P450 CYP2E1, which may result in altered drug metabolism for individuals on certain medications (e.g., chlorzoxazone).[33]
- Tributyltin has been found to inhibit the function of cytochrome P450, leading to masculinization of mollusks.[34]
- Goldenseal, with its two notable alkaloids berberine and hydrastine, has been shown to alter P450-marker enzymatic activities (involving CYP2C9, CYP2D6, and CYP3A4).[35]
Other specific P450 functions edit
Steroid hormones edit

A subset of cytochrome P450 enzymes play important roles in the synthesis of steroid hormones (steroidogenesis) by the adrenals, gonads, and peripheral tissue:
- CYP11A1 (also known as P450scc or P450c11a1) in adrenal mitochondria affects "the activity formerly known as 20,22-desmolase" (steroid 20α-hydroxylase, steroid 22-hydroxylase, cholesterol side-chain scission).
- CYP11B1 (encoding the protein P450c11β) found in the inner mitochondrial membrane of adrenal cortex has steroid 11β-hydroxylase, steroid 18-hydroxylase, and steroid 18-methyloxidase activities.
- CYP11B2 (encoding the protein P450c11AS), found only in the mitochondria of the adrenal zona glomerulosa, has steroid 11β-hydroxylase, steroid 18-hydroxylase, and steroid 18-methyloxidase activities.
- CYP17A1, in endoplasmic reticulum of adrenal cortex has steroid 17α-hydroxylase and 17,20-lyase activities.
- CYP21A2 (P450c21) in adrenal cortex conducts 21-hydroxylase activity.
- CYP19A (P450arom, aromatase) in endoplasmic reticulum of gonads, brain, adipose tissue, and elsewhere catalyzes aromatization of androgens to estrogens.
Polyunsaturated fatty acids and eicosanoids edit
Certain cytochrome P450 enzymes are critical in metabolizing polyunsaturated fatty acids (PUFAs) to biologically active, intercellular cell signaling molecules (eicosanoids) and/or metabolize biologically active metabolites of the PUFA to less active or inactive products. These CYPs possess cytochrome P450 omega hydroxylase and/or epoxygenase enzyme activity.
- CYP1A1, CYP1A2, and CYP2E1 metabolize endogenous PUFAs to signaling molecules: they metabolize arachidonic acid (i.e. AA) to 19-hydroxyeicosatetraenoic acid (i.e. 19-HETE; see 20-hydroxyeicosatetraenoic acid); eicosapentaenoic acid (i.e. EPA) to epoxyeicosatetraenoic acids (i.e. EEQs); and docosahexaenoic acid (i.e. DHA) to epoxydocosapentaenoic acids (i.e. EDPs).
- CYP2C8, CYP2C9, CYP2C18, CYP2C19, and CYP2J2 metabolize endogenous PUFAs to signaling molecules: they metabolize AA to epoxyeicosatetraenoic acids (i.e. EETs); EPA to EEQs; and DHA to EDPs.
- CYP2S1 metabolizes PUFA to signaling molecules: it metabolizes AA to EETs and EPA to EEQs.
- CYP3A4 metabolizes AA to EET signaling molecules.
- CYP4A11 metabolizes endogenous PUFAs to signaling molecules: it metabolizes AA to 20-HETE and EETs; it also hydroxylates DHA to 22-hydroxy-DHA (i.e. 12-HDHA).
- CYP4F2, CYP4F3A, and CYP4F3B (see CYP4F3 for latter two CYPs) metabolize PUFAs to signaling molecules: they metabolizes AA to 20-HETE. They also metabolize EPA to 19-hydroxyeicosapentaenoic acid (19-HEPE) and 20-hydroxyeicosapentaenoic acid (20-HEPE) as well as metabolize DHA to 22-HDA. They also inactivate or reduce the activity of signaling molecules: they metabolize leukotriene B4 (LTB4) to 20-hydroxy-LTB4, 5-hydroxyeicosatetraenoic acid (5-HETE) to 5,20-diHETE, 5-oxo-eicosatetraenoic acid (5-oxo-ETE) to 5-oxo,20-hydroxy-ETE, 12-hydroxyeicosatetraenoic acid (12-HETE) to 12,20-diHETE, EETs to 20-hydroxy-EETs, and lipoxins to 20-hydroxy products.
- CYP4F8 and CYP4F12 metabolize PUFAs to signaling molecules: they metabolizes EPA to EEQs and DHA to EDPs. They also metabolize AA to 18-hydroxyeicosatetraenoic acid (18-HETE) and 19-HETE.
- CYP4F11 inactivates or reduces the activity of signaling molecules: it metabolizes LTB4 to 20-hydroxy-LTB4, (5-HETE) to 5,20-diHETE, (5-oxo-ETE) to 5-oxo,20-hydroxy-ETE, (12-HETE) to 12,20-diHETE, (15-HETE) to 15,20-diHETE, EETs to 20-hydroxy-EETs, and lipoxins to 20-hydroxy products.
- CYP4F22 ω-hydroxylates extremely long "very long chain fatty acids", i.e. fatty acids that are 28 or more carbons long. The ω-hydroxylation of these special fatty acids is critical to creating and maintaining the skin's water barrier function; autosomal recessive inactivating mutations of CYP4F22 are associated with the Lamellar ichthyosis subtype of Congenital ichthyosiform erythrodema in humans.[37]
CYP families in humans edit
Humans have 57 genes and more than 59 pseudogenes divided among 18 families of cytochrome P450 genes and 43 subfamilies.[38] This is a summary of the genes and of the proteins they encode. See the homepage of the cytochrome P450 Nomenclature Committee for detailed information.[20]
| Family | Function | Members | Genes | Pseudogenes |
| CYP1 | drug and steroid (especially estrogen) metabolism, benzo[a]pyrene toxification (forming (+)-benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide) | 3 subfamilies, 3 genes, 1 pseudogene | CYP1A1, CYP1A2, CYP1B1 | CYP1D1P |
| CYP2 | drug and steroid metabolism | 13 subfamilies, 16 genes, 16 pseudogenes | CYP2A6, CYP2A7, CYP2A13, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1, CYP2F1, CYP2J2, CYP2R1, CYP2S1, CYP2U1, CYP2W1 | Too many to list |
| CYP3 | drug and steroid (including testosterone) metabolism | 1 subfamily, 4 genes, 4 pseudogenes | CYP3A4, CYP3A5, CYP3A7, CYP3A43 | CYP3A51P, CYP3A52P, CYP3A54P, CYP3A137P |
| CYP4 | arachidonic acid or fatty acid metabolism | 6 subfamilies, 12 genes, 10 pseudogenes | CYP4A11, CYP4A22, CYP4B1, CYP4F2, CYP4F3, CYP4F8, CYP4F11, CYP4F12, CYP4F22, CYP4V2, CYP4X1, CYP4Z1 | Too many to list |
| CYP5 | thromboxane A2 synthase | 1 subfamily, 1 gene | CYP5A1 | |
| CYP7 | bile acid biosynthesis 7-alpha hydroxylase of steroid nucleus | 2 subfamilies, 2 genes | CYP7A1, CYP7B1 | |
| CYP8 | varied | 2 subfamilies, 2 genes | CYP8A1 (prostacyclin synthase), CYP8B1 (bile acid biosynthesis) | |
| CYP11 | steroid biosynthesis | 2 subfamilies, 3 genes | CYP11A1, CYP11B1, CYP11B2 | |
| CYP17 | steroid biosynthesis, 17-alpha hydroxylase | 1 subfamily, 1 gene | CYP17A1 | |
| CYP19 | steroid biosynthesis: aromatase synthesizes estrogen | 1 subfamily, 1 gene | CYP19A1 | |
| CYP20 | unknown function | 1 subfamily, 1 gene | CYP20A1 | |
| CYP21 | steroid biosynthesis | 1 subfamilies, 1 gene, 1 pseudogene | CYP21A2 | CYP21A1P |
| CYP24 | vitamin D degradation | 1 subfamily, 1 gene | CYP24A1 | |
| CYP26 | retinoic acid hydroxylase | 3 subfamilies, 3 genes | CYP26A1, CYP26B1, CYP26C1 | |
| CYP27 | varied | 3 subfamilies, 3 genes | CYP27A1 (bile acid biosynthesis), CYP27B1 (vitamin D3 1-alpha hydroxylase, activates vitamin D3), CYP27C1 (vitamin A1 to A2) | |
| CYP39 | 7-alpha hydroxylation of 24-hydroxycholesterol | 1 subfamily, 1 gene | CYP39A1 | |
| CYP46 | cholesterol 24-hydroxylase | 1 subfamily, 1 gene, 1 pseudogene | CYP46A1 | CYP46A4P |
| CYP51 | cholesterol biosynthesis | 1 subfamily, 1 gene, 3 pseudogenes | CYP51A1 (lanosterol 14-alpha demethylase) | CYP51P1, CYP51P2, CYP51P3 |
P450s in other species edit
Animals edit
Other animals often have more P450 genes than humans do. Reported numbers range from 35 genes in the sponge Amphimedon queenslandica to 235 genes in the cephalochordate Branchiostoma floridae.[39] Mice have genes for 101 P450s, and sea urchins have even more (perhaps as many as 120 genes).[40] Most CYP enzymes are presumed to have monooxygenase activity, as is the case for most mammalian CYPs that have been investigated (except for, e.g., CYP19 and CYP5). Gene and genome sequencing is far outpacing biochemical characterization of enzymatic function, though many genes with close homology to CYPs with known function have been found, giving clues to their functionality.
The classes of P450s most often investigated in non-human animals are those either involved in development (e.g., retinoic acid or hormone metabolism) or involved in the metabolism of toxic compounds (such as heterocyclic amines or polyaromatic hydrocarbons). Often there are differences in gene regulation or enzyme function of P450s in related animals that explain observed differences in susceptibility to toxic compounds (ex. canines' inability to metabolize xanthines such as caffeine). Some drugs undergo metabolism in both species via different enzymes, resulting in different metabolites, while other drugs are metabolized in one species but excreted unchanged in another species. For this reason, one species's reaction to a substance is not a reliable indication of the substance's effects in humans. A species of Sonoran Desert Drosophila that uses an upregulated expression of the CYP28A1 gene for detoxification of cacti rot is Drosophila mettleri. Flies of this species have adapted an upregulation of this gene due to exposure of high levels of alkaloids in host plants.
P450s have been extensively examined in mice, rats, dogs, and less so in zebrafish, in order to facilitate use of these model organisms in drug discovery and toxicology. Recently P450s have also been discovered in avian species, in particular turkeys, that may turn out to be a useful model for cancer research in humans.[41] CYP1A5 and CYP3A37 in turkeys were found to be very similar to the human CYP1A2 and CYP3A4 respectively, in terms of their kinetic properties as well as in the metabolism of aflatoxin B1.[42]
CYPs have also been extensively studied in insects, often to understand pesticide resistance. For example, CYP6G1 is linked to insecticide resistance in DDT-resistant Drosophila melanogaster[43] and CYP6M2 in the mosquito malaria vector Anopheles gambiae is capable of directly metabolizing pyrethroids.[44] Other cytochromes, such as those in Anopheles gambiae, are under preliminary research for their potential role in pesticide resistance, infectious diseases, and malaria.[45]
Microbial edit
Microbial cytochromes P450 are often soluble enzymes and are involved in diverse metabolic processes. In bacteria the distribution of P450s is very variable with many bacteria having no identified P450s (e.g. E.coli). Some bacteria, predominantly actinomycetes, have numerous P450s (e.g.,[46][47]). Those so far identified are generally involved in either biotransformation of xenobiotic compounds (e.g. CYP105A1 from Streptomyces griseolus metabolizes sulfonylurea herbicides to less toxic derivatives,[48]) or are part of specialised metabolite biosynthetic pathways (e.g. CYP170B1 catalyses production of the sesquiterpenoid albaflavenone in Streptomyces albus[49]). Although no P450 has yet been shown to be essential in a microbe, the CYP105 family is highly conserved with a representative in every streptomycete genome sequenced so far.[50] Due to the solubility of bacterial P450 enzymes, they are generally regarded as easier to work with than the predominantly membrane bound eukaryotic P450s. This, combined with the remarkable chemistry they catalyse, has led to many studies using the heterologously expressed proteins in vitro. Few studies have investigated what P450s do in vivo, what the natural substrate(s) are and how P450s contribute to survival of the bacteria in the natural environment.Three examples that have contributed significantly to structural and mechanistic studies are listed here, but many different families exist.
- Cytochrome P450 cam (CYP101A1) originally from Pseudomonas putida has been used as a model for many cytochromes P450 and was the first cytochrome P450 three-dimensional protein structure solved by X-ray crystallography. This enzyme is part of a camphor-hydroxylating catalytic cycle consisting of two electron transfer steps from putidaredoxin, a 2Fe-2S cluster-containing protein cofactor.
- Cytochrome P450 eryF (CYP107A1) originally from the actinomycete bacterium Saccharopolyspora erythraea is responsible for the biosynthesis of the antibiotic erythromycin by C6-hydroxylation of the macrolide 6-deoxyerythronolide B.
- Cytochrome P450 BM3 (CYP102A1) from the soil bacterium Bacillus megaterium catalyzes the NADPH-dependent hydroxylation of several long-chain fatty acids at the ω–1 through ω–3 positions. Unlike almost every other known CYP (except CYP505A1, cytochrome P450 foxy), it constitutes a natural fusion protein between the CYP domain and an electron donating cofactor. Thus, BM3 is potentially very useful in biotechnological applications.[51][52]
- Cytochrome P450 119 (CYP119A1) isolated from the thermophillic archea Sulfolobus solfataricus [53] has been used in a variety of mechanistic studies.[16] Because thermophillic enzymes evolved to function at high temperatures, they tend to function more slowly at room temperature (if at all) and are therefore excellent mechanistic models.
Fungi edit
The commonly used azole class of antifungal drugs works by inhibition of the fungal cytochrome P450 14α-demethylase.[54][better source needed]
Plants edit
Cytochromes P450 are involved in a variety of processes of plant growth, development, and defense. It is estimated that P450 genes make up approximately 1% of the plant genome.[55][56] These enzymes lead to various fatty acid conjugates, plant hormones, secondary metabolites, lignins, and a variety of defensive compounds.[57]
Cytochromes P450 play an important role in plant defense– involvement in phytoalexin biosynthesis, hormone metabolism, and biosynthesis of diverse secondary metabolites.[58] The expression of cytochrome p450 genes is regulated in response to environmental stresses indicative of a critical role in plant defense mechanisms.[59]
Phytoalexins have shown to be important in plant defense mechanisms as they are antimicrobial compounds produced by plants in response to plant pathogens. Phytoalexins are not pathogen-specific, but rather plant-specific; each plant has its own unique set of phytoalexins. However, they can still attack a wide range of different pathogens. Arabidopsis is a plant closely related to cabbage and mustard and produces the phytoalexin camalexin. Camalexin originates from tryptophan and its biosynthesis involves five cytochrome P450 enzymes. The five cytochrome P450 enzymes include CYP79B2, CYP79B3, CYP71A12, CYP71A13, and CYP71B15. The first step of camalexin biosynthesis produces indole-3-acetaldoxime (IAOx) from tryptophan and is catalyzed by either CYP79B2 or CYP79B3. IAOx is then immediately converted to indole-3-acetonitrile (IAN) and is controlled by either CYP71A13 or its homolog CYP71A12. The last two steps of the biosynthesis pathway of camalexin are catalyzed by CYP71B15. In these steps, indole-3-carboxylic acid (DHCA) is formed from cysteine-indole-3-acetonitrile (Cys(IAN)) followed by the biosynthesis of camalexin. There are some intermediate steps within the pathway that remain unclear, but it is well understood that cytochrome P450 is pivotal in camalexin biosynthesis and that this phytoalexin plays a major role in plant defense mechanisms.[60]
Cytochromes P450 are largely responsible for the synthesis of the jasmonic acid (JA), a common hormonal defenses against abiotic and biotic stresses for plant cells. For example, a P450, CYP74A is involved in the dehydration reaction to produce an insatiable allene oxide from hydroperoxide.[61] JA chemical reactions are critical in the presence of biotic stresses that can be caused by plant wounding, specifically shown in the plant, Arabidopsis. As a prohormone, jasmonic acid must be converted to the JA-isoleucine (JA-Ile) conjugate by JAR1 catalysation in order to be considered activated. Then, JA-Ile synthesis leads to the assembly of the co-receptor complex compo`sed of COI1 and several JAZ proteins. Under low JA-Ile conditions, the JAZ protein components act as transcriptional repressors to suppress downstream JA genes. However, under adequate JA-Ile conditions, the JAZ proteins are ubiquitinated and undergo degradation through the 26S proteasome, resulting in functional downstream effects. Furthermore, several CYP94s (CYP94C1 and CYP94B3) are related to JA-Ile turnover and show that JA-Ile oxidation status impacts plant signaling in a catabolic manner.[55] Cytochrome P450 hormonal regulation in response to extracellular and intracellular stresses is critical for proper plant defense response. This has been proven through thorough analysis of various CYP P450s in jasmonic acid and phytoalexin pathways.
Cytochrome P450 aromatic O-demethylase, which is made of two distinct promiscuous parts: a cytochrome P450 protein (GcoA) and three domain reductase, is significant for its ability to convert Lignin, the aromatic biopolymer common in plant cell walls, into renewable carbon chains in a catabolic set of reactions. In short, it is a facilitator of a critical step in Lignin conversion.
InterPro subfamilies edit
This section may require cleanup to meet Wikipedia's quality standards. The specific problem is: broken links; fragmented paragraph. (September 2016) |
InterPro subfamilies:
- Cytochrome P450, B-class InterPro: IPR002397
- Cytochrome P450, mitochondrial InterPro: IPR002399
- Cytochrome P450, E-class, group I InterPro: IPR002401
- Cytochrome P450, E-class, group II InterPro: IPR002402
- Cytochrome P450, E-class, group IV InterPro: IPR002403
- Aromatase
Clozapine, imipramine, paracetamol, phenacetin Heterocyclic aryl amines Inducible and CYP1A2 5-10% deficient oxidize uroporphyrinogen to uroporphyrin (CYP1A2) in heme metabolism, but they may have additional undiscovered endogenous substrates. are inducible by some polycyclic hydrocarbons, some of which are found in cigarette smoke and charred food.
These enzymes are of interest, because in assays, they can activate compounds to carcinogens. High levels of CYP1A2 have been linked to an increased risk of colon cancer. Since the 1A2 enzyme can be induced by cigarette smoking, this links smoking with colon cancer.[62]
See also edit
References edit
- ^ Gonzalez FJ, Gelboin HV (November 1992). "Human cytochromes P450: evolution and cDNA-directed expression". Environmental Health Perspectives. 98: 81–85. doi:10.1289/ehp.929881. PMC 1519618. PMID 1486867.
- ^ a b "Cytochrome P450". InterPro.
- ^ a b Danielson PB (December 2002). "The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans". Current Drug Metabolism. 3 (6): 561–597. doi:10.2174/1389200023337054. PMID 12369887.
- ^ Lamb DC, Lei L, Warrilow AG, Lepesheva GI, Mullins JG, Waterman MR, Kelly SL (August 2009). "The first virally encoded cytochrome p450". Journal of Virology. 83 (16): 8266–8269. doi:10.1128/JVI.00289-09. PMC 2715754. PMID 19515774.
- ^ Sigel A, Sigel H, Sigel RK (2007). The Ubiquitous Roles of Cytochrome P450 Proteins: Metal Ions in Life Sciences. New York: Wiley. ISBN 978-0-470-01672-5.
- ^ Nelson DR (January 2018). "Cytochrome P450 diversity in the tree of life". Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1866 (1): 141–154. doi:10.1016/j.bbapap.2017.05.003. PMC 5681887. PMID 28502748.
- ^ Nelson DR (October 2009). "The cytochrome p450 homepage". Human Genomics. 4 (1). University of Tennessee: 59–65. doi:10.1186/1479-7364-4-1-59. PMC 3500189. PMID 19951895.
- ^ "NCBI sequence viewer". Retrieved 2007-11-19.
- ^ Nelson DR (October 2009). "The cytochrome p450 homepage". Human Genomics. 4 (1): 59–65. doi:10.1186/1479-7364-4-1-59. PMC 3500189. PMID 19951895.
- ^ Nelson DR (January 2011). "Progress in tracing the evolutionary paths of cytochrome P450". Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1814 (1): 14–18. doi:10.1016/j.bbapap.2010.08.008. PMID 20736090.
- ^ Hanukoglu I (1996). "Electron transfer proteins of cytochrome P450 systems" (PDF). Adv. Mol. Cell Biol. Advances in Molecular and Cell Biology. 14: 29–55. doi:10.1016/S1569-2558(08)60339-2. ISBN 978-0-7623-0113-3.
- ^ [1] Archived 2019-10-18 at the Wayback Machine PROSITE consensus pattern for P450
- ^ a b Meunier B, de Visser SP, Shaik S (September 2004). "Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes". Chemical Reviews. 104 (9): 3947–3980. doi:10.1021/cr020443g. PMID 15352783. S2CID 33927145.
- ^ Poulos TL, Finzel BC, Howard AJ (June 1987). "High-resolution crystal structure of cytochrome P450cam". Journal of Molecular Biology. 195 (3): 687–700. doi:10.1016/0022-2836(87)90190-2. PMID 3656428.
- ^ Sligar SG, Cinti DL, Gibson GG, Schenkman JB (October 1979). "Spin state control of the hepatic cytochrome P450 redox potential". Biochemical and Biophysical Research Communications. 90 (3): 925–932. doi:10.1016/0006-291X(79)91916-8. PMID 228675.
- ^ a b c Rittle J, Green MT (November 2010). "Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics". Science. 330 (6006): 933–937. Bibcode:2010Sci...330..933R. doi:10.1126/science.1193478. PMID 21071661. S2CID 206528205.
- ^ a b Ortiz de Montellano PR (2005). Cytochrome P450: structure, mechanism, and biochemistry (3rd ed.). New York: Kluwer Academic/Plenum Publishers. ISBN 978-0-306-48324-0.
- ^ Hopper CP, Zambrana PN, Goebel U, Wollborn J (June 2021). "A brief history of carbon monoxide and its therapeutic origins". Nitric Oxide. 111–112: 45–63. doi:10.1016/j.niox.2021.04.001. PMID 33838343. S2CID 233205099.
- ^ Berka K, Hendrychová T, Anzenbacher P, Otyepka M (October 2011). "Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site". The Journal of Physical Chemistry A. 115 (41): 11248–11255. Bibcode:2011JPCA..11511248B. doi:10.1021/jp204488j. PMC 3257864. PMID 21744854.
- ^ a b "P450 Table". 8 April 2021.
- ^ doctorfungus > Antifungal Drug Interactions Archived 2012-08-01 at archive.today Content Director: Russell E. Lewis, Pharm.D. Retrieved on Jan 23, 2010
- ^ Guengerich FP (January 2008). "Cytochrome p450 and chemical toxicology". Chemical Research in Toxicology. 21 (1): 70–83. doi:10.1021/tx700079z. PMID 18052394. S2CID 17548932. (Metabolism in this context is the chemical modification or degradation of drugs.)
- ^ Bains RK (2013). "African variation at Cytochrome P450 genes". Evolution, Medicine, and Public Health. 2013 (1): 118–134. doi:10.1093/emph/eot010. ISSN 2050-6201. PMC 3868406. PMID 24481193.
- ^ Hodges RE, Minich DM (2015). "Modulation of Metabolic Detoxification Pathways Using Foods and Food-Derived Components: A Scientific Review with Clinical Application". Journal of Nutrition and Metabolism. 2015: 1–23. doi:10.1155/2015/760689. ISSN 2090-0724. PMC 4488002. PMID 26167297.
- ^ Zahno A, Brecht K, Morand R, Maseneni S, Török M, Lindinger PW, Krähenbühl S (February 2011). "The role of CYP3A4 in amiodarone-associated toxicity on HepG2 cells". Biochemical Pharmacology. 81 (3): 432–441. doi:10.1016/j.bcp.2010.11.002. PMID 21070748.
- ^ "Carbamazepine: Watch for Many Potential Drug Interactions". Pharmacy Times. Archived from the original on 2020-10-14. Retrieved 2019-11-07.
- ^ Bailey DG, Dresser GK (2004). "Interactions between grapefruit juice and cardiovascular drugs". American Journal of Cardiovascular Drugs. 4 (5): 281–297. doi:10.2165/00129784-200404050-00002. PMID 15449971. S2CID 11525439.
- ^ Zeratsky K (2008-11-06). "Grapefruit juice: Can it cause drug interactions?". Ask a food & nutrition specialist. MayoClinic.com. Retrieved 2009-02-09.
- ^ Chaudhary A, Willett KL (January 2006). "Inhibition of human cytochrome CYP 1 enzymes by flavonoids of St. John's wort". Toxicology. 217 (2–3): 194–205. doi:10.1016/j.tox.2005.09.010. PMID 16271822.
- ^ Strandell J, Neil A, Carlin G (February 2004). "An approach to the in vitro evaluation of potential for cytochrome P450 enzyme inhibition from herbals and other natural remedies". Phytomedicine. 11 (2–3): 98–104. doi:10.1078/0944-7113-00379. PMID 15070158.
- ^ Kroon LA (September 2007). "Drug interactions with smoking". American Journal of Health-System Pharmacy. 64 (18): 1917–1921. doi:10.2146/ajhp060414. PMID 17823102. S2CID 5397510.
- ^ Zhang JW, Liu Y, Cheng J, Li W, Ma H, Liu HT, et al. (2007). "Inhibition of human liver cytochrome P450 by star fruit juice". Journal of Pharmacy & Pharmaceutical Sciences. 10 (4): 496–503. doi:10.18433/j30593. PMID 18261370.
- ^ Leclercq I, Desager JP, Horsmans Y (August 1998). "Inhibition of chlorzoxazone metabolism, a clinical probe for CYP2E1, by a single ingestion of watercress". Clinical Pharmacology and Therapeutics. 64 (2): 144–149. doi:10.1016/S0009-9236(98)90147-3. PMID 9728894. S2CID 43863786.
- ^ Walmsley S. "Tributyltin pollution on a global scale. An overview of relevant and recent research: impacts and issues" (PDF). WWF UK. Archived from the original (PDF) on 2014-04-07. Retrieved 2014-05-01.
- ^ Chatterjee P, Franklin MR (November 2003). "Human cytochrome p450 inhibition and metabolic-intermediate complex formation by goldenseal extract and its methylenedioxyphenyl components". Drug Metabolism and Disposition. 31 (11): 1391–1397. doi:10.1124/dmd.31.11.1391. PMID 14570772. S2CID 2967171.
- ^ Häggström M, Richfield D (2014). "Diagram of the pathways of human steroidogenesis". WikiJournal of Medicine. 1 (1). doi:10.15347/wjm/2014.005. ISSN 2002-4436.
- ^ Sugiura K, Akiyama M (July 2015). "Update on autosomal recessive congenital ichthyosis: mRNA analysis using hair samples is a powerful tool for genetic diagnosis". Journal of Dermatological Science. 79 (1): 4–9. doi:10.1016/j.jdermsci.2015.04.009. PMID 25982146.
- ^ Nelson D (2003). Cytochromes P450 in humans. Retrieved May 9, 2005.
- ^ Nelson DR, Goldstone JV, Stegeman JJ (February 2013). "The cytochrome P450 genesis locus: the origin and evolution of animal cytochrome P450s". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 368 (1612): 20120474. doi:10.1098/rstb.2012.0474. PMC 3538424. PMID 23297357.
- ^ Goldstone JV, Hamdoun A, Cole BJ, Howard-Ashby M, Nebert DW, Scally M, et al. (December 2006). "The chemical defensome: environmental sensing and response genes in the Strongylocentrotus purpuratus genome". Developmental Biology. 300 (1): 366–384. doi:10.1016/j.ydbio.2006.08.066. PMC 3166225. PMID 17097629.
- ^ Rawal S, Kim JE, Coulombe R (December 2010). "Aflatoxin B1 in poultry: toxicology, metabolism and prevention". Research in Veterinary Science. 89 (3): 325–331. doi:10.1016/j.rvsc.2010.04.011. PMID 20462619.
- ^ Rawal S, Coulombe RA (August 2011). "Metabolism of aflatoxin B1 in turkey liver microsomes: the relative roles of cytochromes P450 1A5 and 3A37". Toxicology and Applied Pharmacology. 254 (3): 349–354. doi:10.1016/j.taap.2011.05.010. PMID 21616088.
- ^ McCart C, Ffrench-Constant RH (June 2008). "Dissecting the insecticide-resistance- associated cytochrome P450 gene Cyp6g1". Pest Management Science. 64 (6): 639–645. doi:10.1002/ps.1567. PMID 18338338. S2CID 41480564.
- ^ Ismail HM, O'Neill PM, Hong DW, Finn RD, Henderson CJ, Wright AT, et al. (December 2013). "Pyrethroid activity-based probes for profiling cytochrome P450 activities associated with insecticide interactions". Proceedings of the National Academy of Sciences of the United States of America. 110 (49): 19766–19771. Bibcode:2013PNAS..11019766I. doi:10.1073/pnas.1320185110. PMC 3856776. PMID 24248381.
- ^ Skorokhod O, Vostokova E, Gilardi G (2024). "The role of P450 enzymes in malaria and other vector-borne infectious diseases". Biofactors. 50 (1): 16–32. doi:10.1002/biof.1996. PMID 37555735.
- ^ McLean KJ, Clift D, Lewis DG, Sabri M, Balding PR, Sutcliffe MJ, et al. (May 2006). "The preponderance of P450s in the Mycobacterium tuberculosis genome". Trends in Microbiology. 14 (5): 220–228. doi:10.1016/j.tim.2006.03.002. PMID 16581251.
- ^ Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, et al. (May 2003). "Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis". Nature Biotechnology. 21 (5): 526–531. doi:10.1038/nbt820. PMID 12692562.
- ^ O'Keefe DP, Romesser JA, Leto KJ (1988). "Identification of constitutive and herbicide inducible cytochromes P-450 in Streptomyces griseolus". Archives of Microbiology. 149 (5). Springer Science+Business: 406–412. Bibcode:1988ArMic.149..406O. doi:10.1007/bf00425579. ISSN 0302-8933. S2CID 35526991.
- ^ Moody SC, Zhao B, Lei L, Nelson DR, Mullins JG, Waterman MR, et al. (May 2012). "Investigating conservation of the albaflavenone biosynthetic pathway and CYP170 bifunctionality in streptomycetes". The FEBS Journal. 279 (9): 1640–1649. doi:10.1111/j.1742-4658.2011.08447.x. PMID 22151149.
- ^ Moody SC, Loveridge EJ (December 2014). "CYP105-diverse structures, functions and roles in an intriguing family of enzymes in Streptomyces". Journal of Applied Microbiology. 117 (6): 1549–1563. doi:10.1111/jam.12662. PMC 4265290. PMID 25294646.
- ^ Narhi LO, Fulco AJ (June 1986). "Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium". The Journal of Biological Chemistry. 261 (16): 7160–7169. doi:10.1016/S0021-9258(17)38369-2. PMID 3086309.
- ^ Girvan HM, Waltham TN, Neeli R, Collins HF, McLean KJ, Scrutton NS, et al. (December 2006). "Flavocytochrome P450 BM3 and the origin of CYP102 fusion species". Biochemical Society Transactions. 34 (Pt 6): 1173–1177. doi:10.1042/BST0341173. PMID 17073779.
- ^ Wright RL, Harris K, Solow B, White RH, Kennelly PJ (April 1996). "Cloning of a potential cytochrome P450 from the archaeon Sulfolobus solfataricus". FEBS Letters. 384 (3): 235–239. doi:10.1016/0014-5793(96)00322-5. PMID 8617361. S2CID 19579406.
- ^ Vanden Bossche H, Marichal P, Gorrens J, Coene MC (September 1990). "Biochemical basis for the activity and selectivity of oral antifungal drugs". British Journal of Clinical Practice. Supplement. 71: 41–46. PMID 2091733.
- ^ a b Mizutani M (2012). "Impacts of diversification of cytochrome P450 on plant metabolism". Biological & Pharmaceutical Bulletin. 35 (6): 824–832. doi:10.1248/bpb.35.824. PMID 22687470.
- ^ Mizutani M, Sato F (March 2011). "Unusual P450 reactions in plant secondary metabolism". Archives of Biochemistry and Biophysics. P450 Catalysis Mechanisms. 507 (1): 194–203. doi:10.1016/j.abb.2010.09.026. PMID 20920462.
- ^ Schuler MA, Werck-Reichhart D (2003-01-01). "Functional genomics of P450s". Annual Review of Plant Biology. 54 (1): 629–667. doi:10.1146/annurev.arplant.54.031902.134840. PMID 14503006.
- ^ Xu J, Wang X, Guo W (2015-09-01). "The cytochrome P450 superfamily: Key players in plant development and defense". Journal of Integrative Agriculture. 14 (9): 1673–1686. doi:10.1016/S2095-3119(14)60980-1. ISSN 2095-3119.
- ^ Heitz T, Widemann E, Lugan R, Miesch L, Ullmann P, Désaubry L, et al. (February 2012). "Cytochromes P450 CYP94C1 and CYP94B3 catalyze two successive oxidation steps of plant hormone Jasmonoyl-isoleucine for catabolic turnover". The Journal of Biological Chemistry. 287 (9): 6296–6306. doi:10.1074/jbc.M111.316364. PMC 3307330. PMID 22215670.
- ^ Pandian BA, Sathishraj R, Djanaguiraman M, Prasad PV, Jugulam M (May 2020). "Role of Cytochrome P450 Enzymes in Plant Stress Response". Antioxidants. 9 (5): 454. doi:10.3390/antiox9050454. PMC 7278705. PMID 32466087.
- ^ "Canvas Login". login.canvas.uw.edu. Retrieved 2022-06-07.
- ^ Petros WP, Younis IR, Ford JN, Weed SA (October 2012). "Effects of tobacco smoking and nicotine on cancer treatment". Pharmacotherapy. 32 (10): 920–931. doi:10.1002/j.1875-9114.2012.01117. PMC 3499669. PMID 23033231.
Further reading edit
- Gelboin HV, Krausz K (March 2006). "Monoclonal antibodies and multifunctional cytochrome P450: drug metabolism as paradigm". Journal of Clinical Pharmacology. 46 (3): 353–372. doi:10.1177/0091270005285200. PMID 16490812. S2CID 33325397.
- Gelboin HV, Krausz KW, Gonzalez FJ, Yang TJ (November 1999). "Inhibitory monoclonal antibodies to human cytochrome P450 enzymes: a new avenue for drug discovery". Trends in Pharmacological Sciences. 20 (11): 432–438. doi:10.1016/S0165-6147(99)01382-6. PMID 10542439.
- "Cytochrome P450 Mediated Drug and Carcinogen Metabolism using Monoclonal Antibodies". home.ccr.cancer.gov. Retrieved 2018-04-02.
- Krausz KW, Goldfarb I, Buters JT, Yang TJ, Gonzalez FJ, Gelboin HV (November 2001). "Monoclonal antibodies specific and inhibitory to human cytochromes P450 2C8, 2C9, and 2C19". Drug Metabolism and Disposition. 29 (11): 1410–1423. PMID 11602516.
- Gonzalez FJ, Gelboin HV (1994). "Role of human cytochromes P450 in the metabolic activation of chemical carcinogens and toxins". Drug Metabolism Reviews. 26 (1–2): 165–183. doi:10.3109/03602539409029789. PMID 8082563.
- Estabrook RW (December 2003). "A passion for P450s (Remembrances of the early history of research on cytochrome P450)". Drug Metabolism and Disposition. 31 (12): 1461–1473. doi:10.1124/dmd.31.12.1461. PMID 14625342. S2CID 43655270.
External links edit
- Degtyarenko K (2009-01-09). "Directory of P450-containing Systems". International Centre for Genetic Engineering and Biotechnology. Archived from the original on 2016-07-16. Retrieved 2009-02-10.
- Flockhart DA (2008-09-04). "Human Cytochrome P450 (CYP) Allele Nomenclature Committee". Karolinska Institutet. Retrieved 2009-02-10.
- Preissner S (2010). "Cytochrome P450 database". Nucleic Acids Research. Archived from the original on 2011-11-03. Retrieved 2011-08-02.
- Sigaroudi A, Vollbrecht H (2019). "pharmacokinetic interaction table". Sigaroudi & Vollbrecht.
- Sim SC (2007). "Cytochrome P450 drug interaction table". Indiana University-Purdue University Indianapolis. Retrieved 2009-02-10.