In nuclear magnetic resonance (NMR) spectroscopy, the chemical shift is the resonant frequency of an atomic nucleus relative to a standard in a magnetic field. Often the position and number of chemical shifts are diagnostic of the structure of a molecule.[1][2][3] Chemical shifts are also used to describe signals in other forms of spectroscopy such as photoemission spectroscopy.
Some atomic nuclei possess a magnetic moment (nuclear spin), which gives rise to different energy levels and resonance frequencies in a magnetic field. The total magnetic field experienced by a nucleus includes local magnetic fields induced by currents of electrons in the molecular orbitals (electrons have a magnetic moment themselves). The electron distribution of the same type of nucleus (e.g. 1H, 13C, 15N) usually varies according to the local geometry (binding partners, bond lengths, angles between bonds, and so on), and with it the local magnetic field at each nucleus. This is reflected in the spin energy levels (and resonance frequencies). The variations of nuclear magnetic resonance frequencies of the same kind of nucleus, due to variations in the electron distribution, is called the chemical shift. The size of the chemical shift is given with respect to a reference frequency or reference sample (see also chemical shift referencing), usually a molecule with a barely distorted electron distribution.
Operating frequency edit
The operating (or Larmor) frequency of a magnet (usually quoted as absolute value in MHz) is calculated from the Larmor equation[4]


where B0 is the induction of the magnet (SI units of Tesla), and is the magnetogyric ratio of the nucleus — an empirically measured fundamental constant determined by the details of the structure of each nucleus. For example, the proton operating frequency for a 1 Tesla magnet is calculated as:


MRI scanners are often referred to by their field strengths B0 (e.g. "a 7 T scanner"), whereas NMR spectrometers are commonly referred to by the corresponding proton Larmor frequency (e.g. "a 300 MHz spectrometer", which has a B0 of 7 T ). While chemical shift is referenced in order that the units are equivalent across different field strengths, the actual frequency separation in Hertz scales with field strength (B0). As a result, the difference of chemical shift between two signals (ppm) represents a larger number of Hertz on machines that have larger B0 and therefore the signals are less likely to be overlapping in the resulting spectrum. This increased resolution is a significant advantage for analysis. (Larger field machines are also favoured on account of having intrinsically higher signal arising from the Boltzmann distribution of magnetic spin states.)
Chemical shift referencing edit
Chemical shift δ is usually expressed in parts per million (ppm) by frequency, because it is calculated from:[5]

where νsample is the absolute resonance frequency of the sample and νref is the absolute resonance frequency of a standard reference compound, measured in the same applied magnetic field B0. Since the numerator is usually expressed in hertz, and the denominator in megahertz, δ is expressed in ppm.
The detected frequencies (in Hz) for 1H, 13C, and 29Si nuclei are usually referenced against TMS (tetramethylsilane), TSP (Trimethylsilylpropanoic acid), or DSS, which by the definition above have a chemical shift of zero if chosen as the reference. Other standard materials are used for setting the chemical shift for other nuclei.
Thus, an NMR signal observed at a frequency 300 Hz higher than the signal from TMS, where the TMS resonance frequency is 300 MHz, has a chemical shift of:

Although the absolute resonance frequency depends on the applied magnetic field, the chemical shift is independent of external magnetic field strength. On the other hand, the resolution of NMR will increase with applied magnetic field.
Referencing methods edit
Practically speaking, diverse methods may be used to reference chemical shifts in an NMR experiment, which can be subdivided into indirect and direct referencing methods.[5] Indirect referencing uses a channel other than the one of interest to adjust chemical shift scale correctly, i.e. the solvent signal in the deuterium (lock) channel can be used to reference the a 1H NMR spectrum.[5] Both indirect and direct referencing can be done as three different procedures:
- Internal referencing, where the reference compound is added directly to the system under study."[5] In this common practice, users adjust residual solvent signals of 1H or 13C NMR spectra with calibrated spectral tables.[6][7] If substances other than the solvent itself are used for internal referencing, the sample has to be combined with the reference compound, which may affect the chemical shifts.
- External referencing, involving sample and reference contained separately in coaxial cylindrical tubes."[5] With this procedure, the reference signal is still visible in the spectrum of interest, although the reference and the sample are physically separated by a glass wall. Magnetic susceptibility differences between the sample and the reference phase need to be corrected theoretically,[5] which lowers the practicality of this procedure.
- Substitution method: The use of separate cylindrical tubes for the sample and the reference compound, with (in principle) spectra recorded individually for each."[5] Similar to external referencing, this method allows referencing without sample contamination. If field/frequency locking via the 2H signal of the deuterated solvent is used and the solvents of reference and analyte are the same, the use of this methods is straightforward. Problems may arise if different solvents are used for the reference compound and the sample as (just like for external referencing) magnetic susceptibility differences need to be corrected theoretically.[5][8] If this method is used without field/frequency locking, shimming procedures between the sample and the reference need to be avoided as they change the applied magnetic field (and thereby influence the chemical shift).[5]
Modern NMR spectrometers commonly make use of the absolute scale,[8][5] which defines the 1H signal of TMS as 0 ppm in proton NMR and the center frequencies of all other nuclei as percentage of the TMS resonance frequency:[5][8]
![{\displaystyle \Xi [\%]=100(\upsilon _{X}^{obs}/\upsilon _{TMS}^{obs})}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6f20c591d5c692003a4db4745c5e153f08c11db2)
The use of the deuterium (lock) channel, so the 2H signal of the deuterated solvent, and the Ξ value of the absolute scale is a form of internal referencing and is particularly useful in heteronuclear NMR spectroscopy as local reference compounds may not be always be available or easily used (i.e. liquid NH3 for 15N NMR spectroscopy). This system, however, relies on accurately determined 2H NMR chemical shifts enlisted in the spectrometer software and correctly determined Ξ values by IUPAC.[5][8] A recent study for 19F NMR spectroscopy revealed that the use of the absolute scale and lock-based internal referencing led to errors in chemical shifts.[9][10] These may be negated by inclusion of calibrated reference compounds.[9][10]
The induced magnetic field edit
The electrons around a nucleus will circulate in a magnetic field and create a secondary induced magnetic field. This field opposes the applied field as stipulated by Lenz's law and atoms with higher induced fields (i.e., higher electron density) are therefore called shielded, relative to those with lower electron density. Electron-donating alkyl groups, for example, lead to increased shielding whereas electron-withdrawing substituents such as nitro groups lead to deshielding of the nucleus. Not only substituents cause local induced fields. Bonding electrons can also lead to shielding and deshielding effects. A striking example of this is the pi bonds in benzene. Circular current through the hyperconjugated system causes a shielding effect at the molecule's center and a deshielding effect at its edges. Trends in chemical shift are explained based on the degree of shielding or deshielding.
Nuclei are found to resonate in a wide range to the left (or more rare to the right) of the internal standard. When a signal is found with a higher chemical shift:
- the applied effective magnetic field is lower, if the resonance frequency is fixed (as in old traditional CW spectrometers)
- the frequency is higher, when the applied magnetic field is static (normal case in FT spectrometers)
- the nucleus is more deshielded
- the signal or shift is downfield or at low field or paramagnetic
Conversely a lower chemical shift is called a diamagnetic shift, and is upfield and more shielded.
Diamagnetic shielding edit
In real molecules protons are surrounded by a cloud of charge due to adjacent bonds and atoms. In an applied magnetic field (B0) electrons circulate and produce an induced field (Bi) which opposes the applied field. The effective field at the nucleus will be B = B0 − Bi. The nucleus is said to be experiencing a diamagnetic shielding.
Factors causing chemical shifts edit
Important factors influencing chemical shift are electron density, electronegativity of neighboring groups and anisotropic induced magnetic field effects.
Electron density shields a nucleus from the external field. For example, in proton NMR the electron-poor tropylium ion has its protons downfield at 9.17 ppm, those of the electron-rich cyclooctatetraenyl anion move upfield to 6.75 ppm and its dianion even more upfield to 5.56 ppm.
A nucleus in the vicinity of an electronegative atom experiences reduced electron density and the nucleus is therefore deshielded. In proton NMR of methyl halides (CH3X) the chemical shift of the methyl protons increase in the order I < Br < Cl < F from 2.16 ppm to 4.26 ppm reflecting this trend. In carbon NMR the chemical shift of the carbon nuclei increase in the same order from around −10 ppm to 70 ppm. Also when the electronegative atom is removed further away the effect diminishes until it can be observed no longer.
Anisotropic induced magnetic field effects are the result of a local induced magnetic field experienced by a nucleus resulting from circulating electrons that can either be paramagnetic when it is parallel to the applied field or diamagnetic when it is opposed to it. It is observed in alkenes where the double bond is oriented perpendicular to the external field with pi electrons likewise circulating at right angles. The induced magnetic field lines are parallel to the external field at the location of the alkene protons which therefore shift downfield to a 4.5 ppm to 7.5 ppm range. The three-dimensional space where a diamagnetic shift is called the shielding zone with a cone-like shape aligned with the external field.
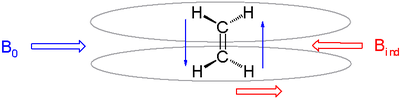
Induced magnetic field of alkenes in external magnetic fields, field lines in grey.

The protons in aromatic compounds are shifted downfield even further with a signal for benzene at 7.73 ppm as a consequence of a diamagnetic ring current.
Alkyne protons by contrast resonate at high field in a 2–3 ppm range. For alkynes the most effective orientation is the external field in parallel with electrons circulation around the triple bond. In this way the acetylenic protons are located in the cone-shaped shielding zone hence the upfield shift.

Induced magnetic field of alkynes in external magnetic fields, field lines in grey.

Magnetic properties of most common nuclei edit
1H and 13C are not the only nuclei susceptible to NMR experiments. A number of different nuclei can also be detected, although the use of such techniques is generally rare due to small relative sensitivities in NMR experiments (compared to 1H) of the nuclei in question, the other factor for rare use being their slender representation in nature and organic compounds.
| Isotope | Occurrence in nature (%) |
Spin number I | Magnetic moment μ (μN) |
Electric quadrupole moment (e × 10−24 cm2) |
Operating frequency at 7 T (MHz) |
Relative sensitivity |
|---|---|---|---|---|---|---|
| 1H | 99.984 | 1/2 | 2.79628 | 0 | 300.13 | 1 |
| 2H | 0.016 | 1 | 0.85739 | 0.0028 | 46.07 | 0.0964 |
| 10B | 18.8 | 3 | 1.8005 | 0.074 | 32.25 | 0.0199 |
| 11B | 81.2 | 3/2 | 2.6880 | 0.026 | 96.29 | 0.165 |
| 12C | 98.9 | 0 | 0 | 0 | 0 | 0 |
| 13C | 1.1 | 1/2 | 0.70220 | 0 | 75.47 | 0.0159 |
| 14N | 99.64 | 1 | 0.40358 | 0.071 | 21.68 | 0.00101 |
| 15N | 0.37 | 1/2 | −0.28304 | 0 | 30.41 | 0.00104 |
| 16O | 99.76 | 0 | 0 | 0 | 0 | 0 |
| 17O | 0.0317 | 5/2 | −1.8930 | −0.0040 | 40.69 | 0.0291 |
| 19F | 100 | 1/2 | 2.6273 | 0 | 282.40 | 0.834 |
| 28Si | 92.28 | 0 | 0 | 0 | 0 | 0 |
| 29Si | 4.70 | 1/2 | −0.5548 | 0 | 59.63 | 0.0785 |
| 31P | 100 | 1/2 | 1.1205 | 0 | 121.49 | 0.0664 |
| 35Cl | 75.4 | 3/2 | 0.92091 | −0.079 | 29.41 | 0.0047 |
| 37Cl | 24.6 | 3/2 | 0.68330 | −0.062 | 24.48 | 0.0027 |
1H, 13C, 15N, 19F and 31P are the five nuclei that have the greatest importance in NMR experiments:
- 1H because of high sensitivity and vast occurrence in organic compounds
- 13C because of being the key component of all organic compounds despite occurring at a low abundance (1.1%) compared to the major isotope of carbon 12C, which has a spin of 0 and therefore is NMR-inactive.
- 15N because of being a key component of important biomolecules such as proteins and DNA
- 19F because of high relative sensitivity
- 31P because of frequent occurrence in organic compounds and moderate relative sensitivity
Chemical shift manipulation edit
In general, the associated increased signal-to-noise and resolution has driven a move towards increasingly high field strengths. In limited cases, however, lower fields are preferred; examples are for systems in chemical exchange, where the speed of the exchange relative to the NMR experiment can cause additional and confounding linewidth broadening. Similarly, while avoidance of second order coupling is generally preferred, this information can be useful for elucidation of chemical structures. Using refocussing pulses placed between recording of successive points of the free induction decay, in an analogous fashion to the spin echo technique in MRI, the chemical shift evolution can be scaled to provide apparent low-field spectra on a high-field spectrometer.[12] In a similar fashion, it is possible to upscale the effect of J-coupling relative to the chemical shift using pulse sequences that include additional J-coupling evolution periods interspersed with conventional spin evolutions.[13]
Other chemical shifts edit
The Knight shift (first reported in 1949) and Shoolery's rule are observed with pure metals and methylene groups, respectively. The NMR chemical shift in its present-day meaning first appeared in journals in 1950. Chemical shifts with a different meaning appear in X-ray photoelectron spectroscopy as the shift in atomic core-level energy due to a specific chemical environment. The term is also used in Mössbauer spectroscopy, where similarly to NMR it refers to a shift in peak position due to the local chemical bonding environment. As is the case for NMR the chemical shift reflects the electron density at the atomic nucleus.[14]
See also edit
- EuFOD, a shift agent
- MRI
- Nuclear magnetic resonance
- Nuclear magnetic resonance spectroscopy of carbohydrates
- Nuclear magnetic resonance spectroscopy of nucleic acids
- Nuclear magnetic resonance spectroscopy of proteins
- Protein NMR
- Random coil index
- Relaxation (NMR)
- Solid-state NMR
- TRISPHAT, a chiral shift reagent for cations
- Zeeman effect
References edit
- ^ Silverstein; Bassler; Morrill (1981). Spectrometric Identification of organic Compounds (4th ed.). ISBN 978-0-471-09070-0.
- ^ Kemp, William (1987). Organic Spectroscopy (3rd ed.). ISBN 978-0-333-41767-6.
- ^ Balei, Metin. Basic 1H and 13C-NMR spectroscopy. ISBN 978-0-444-51811-8.
- ^ "Chemical Shift". NMRCentral. Archived from the original on 26 September 2011.
- ^ a b c d e f g h i j k l Harris, R. K.; Becker, E. D.; Cabral de Menezes, S. M.; Goodfellow, R.; Granger, P. (2001). "NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001)". Pure Appl. Chem. 73 (11): 1795–1818. doi:10.1351/pac200173111795.
- ^ Gottlieb, Hugo E.; Kotlyar, Vadim; Nudelman, Abraham (1997). "NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities". The Journal of Organic Chemistry. 62 (21): 7512–7515. doi:10.1021/jo971176v. ISSN 0022-3263. PMID 11671879.
- ^ Fulmer, Gregory R.; Miller, Alexander J. M.; Sherden, Nathaniel H.; Gottlieb, Hugo E.; Nudelman, Abraham; Stoltz, Brian M.; Bercaw, John E.; Goldberg, Karen I. (10 May 2010). "NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist" (PDF). Organometallics. 29 (9): 2176–2179. doi:10.1021/om100106e. ISSN 0276-7333.
- ^ a b c d Harris, Robin K.; Becker, Edwin D.; Menezes, Cabral de; M, Sonia; Granger, Pierre; Hoffman, Roy E.; Zilm, Kurt W. (2008). "Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008)". Pure and Applied Chemistry. 80 (1): 59–84. doi:10.1351/pac200880010059. ISSN 0033-4545.
- ^ a b Rosenau, Carl Philipp; Jelier, Benson J.; Gossert, Alvar D.; Togni, Antonio (16 May 2018). "Exposing the Origins of Irreproducibility in Fluorine NMR Spectroscopy". Angewandte Chemie International Edition. 51 (30): 9528–9533. doi:10.1002/anie.201802620. ISSN 1433-7851. PMID 29663671.
- ^ a b Rosenau, Carl Philipp; Jelier, Benson J.; Gossert, Alvar D.; Togni, Antonio (16 May 2018). "Fluor-NMR-Spektroskopie rekalibriert". Angewandte Chemie (in German). 130 (30): 9672–9677. Bibcode:2018AngCh.130.9672R. doi:10.1002/ange.201802620. ISSN 0044-8249.
- ^ CRC Handbook of Chemistry and Physics (65th ed.).
- ^ Morris, Gareth A.; Jerome, Neil P.; Lian, Lu-Yun (17 February 2003). "Real‐Time Chemical‐Shift Scaling in High‐Resolution NMR Spectroscopy". Angewandte Chemie (in German). 115 (7): 847–849. doi:10.1002/ange.200390189.
- ^ Glanzer, Simon; Zangger, Klaus (13 April 2015). "Visualizing Unresolved Scalar Couplings by Real-Time-Upscaled NMR". Journal of the American Chemical Society. 137 (15): 5163–5169. doi:10.1021/jacs.5b01687. PMC 4415032. PMID 25837306.
- ^ Nagaoka, Shin'ichi (May 2007). "A Short History of Three Chemical Shifts". J. Chem. Educ. 84 (5): 801. Bibcode:2007JChEd..84..801N. doi:10.1021/ed084p801.
External links edit
- chem.wisc.edu
- BioMagResBank
- NMR Table
- Proton chemical shifts
- Carbon chemical shifts
- Online tutorials (these generally involve combined use of IR, 1H NMR, 13C NMR and mass spectrometry)
- Problem set 1 (see also this link for more background information on spin-spin coupling)
- Problem set 2
- Problem set 4
- Problem set 5
- Combined solutions to problem set 5 (Problems 1–32) and (Problems 33–64)
{kind=link}
{kind=link}