Epilepsy syndromes edit
The most widespread classification of the epilepsies[1] divides epilepsy syndromes by location or distribution of seizures (as revealed by the appearance of the seizures and by EEG) and by cause. Syndromes are divided into localization-related epilepsies, generalized epilepsies, or epilepsies of unknown localization.
Localization-related epilepsies, sometimes termed partial or focal epilepsies, arise from an epileptic focus, a small portion of the brain that serves as the irritant driving the epileptic response. Generalized epilepsies, in contrast, arise from many independent foci (multifocal epilepsies) or from epileptic circuits that involve the whole brain. Epilepsies of unknown localization remain unclear whether they arise from a portion of the brain or from more widespread circuits.
Epilepsy syndromes are further divided by presumptive cause: idiopathic, symptomatic, and cryptogenic. Idiopathic epilepsies are generally thought to arise from genetic abnormalities that lead to alteration of basic neuronal regulation. Symptomatic epilepsies arise from the effects of an epileptic lesion, whether that lesion is focal, such as a tumor, or a defect in metabolism causing widespread injury to the brain. Cryptogenic epilepsies involve a presumptive lesion that is otherwise difficult or impossible to uncover during evaluation.
Some epileptic syndromes are difficult to fit within this classification scheme and fall in the unknown localization/etiology catagory. People who only have had a single seizure, or those with seizures that occur only after specific precipitants ("provoked seizures"), have "epilepsies" that fall into this catagory. Febrile convulsions are an example of seizures bound to a particular precipitant. Landau-Kleffner syndrome is another epilepsy which, because of its variety of EEG distributions, falls uneasily in clear catagories. More confusingly, certain syndromes like West syndrome featuring seizures such as Infantile spasms can be classified as idiopathic, syndromic, or cryptogenic depending on cause and can arise from both focal or generalized epileptic lesions.
- Benign centrotemporal lobe epilepsy of childhood or Benign rolandic epilepsy is an idiopathic localization-related epilepsy that occurs in children between the ages of 3 and 13 years with peak onset in prepubertal late childhood. Apart from their seizure disorder, these patients are otherwise normal. This syndrome features simple partial seizures that involve facial muscles and frequently cause drooling. Although most episodes are brief, seizures sometimes spread and generalize. Seizures are typically nocturnal and confined to sleep. The EEG may demonstrate spike discharges that occur over the centrotemporal scalp over the central sulcus of the brain (the Rolandic sulcus) that are predisposed to occur during drowsiness or light sleep. Seizures cease near puberty.[2] Seizures may require anticonvulsant treatment, but sometimes are infrequent enough to allow physicians to defer treatment.
- Benign occipital epilepsy of childhood (BOEC) is an idiopathic localization-related epilepsy and consists of an evolving group of syndromes. Most authorities include two subtypes, an early subtype with onset between 3-5 years and an late onset between 7-10 years. Seizures in BOEC usually feature visual symptoms such as scotoma or fortifications (brightly colored spots or lines) or amaurosis (blindness or impairment of vision). Convulsions involving one half the body, hemiconvulsions, or forced eye deviation or head turning are common. Younger patients typically experience symptoms similar to migraine with nausea and headache, and older patients typically complain of more visual symptoms. The EEG in BOEC shows spikes recorded from the occipital (back of head) regions. Lately, a group of epilepsies termed Panayiotopolous syndrome[3] that share some clinical features of BOEC but have a wider variety of EEG findings are classified by some as BOEC.
- Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is an idiopathic localization-related epilepsy that is an inheirited epileptic disorder that causes seizures during sleep. Onset is usually in childhood. These seizures arise from the frontal lobes and consist of complex motor movements, such as hand clenching, arm raising/lowering, and knee bending. Vocalizations such as shouting, moaning, or crying are also common. ADNFLE is often misdiagnosed as nightmares. ADNFLE has a genetic basis[4]. These genes encode various nicotinic acetylcholine receptors.
- Primary reading epilepsy is a reflex epilepsy classified as an idiopathic localization-related epilepsy. Reading in susceptible individuals triggers characteristic seizures.
- Childhood absence epilepsy (CEA) affects children between the ages of 4 and 12 years of age, although peak onset is around 5-6 years old. These patients have recurrent absence seizures, brief episodes of unresponsive staring, sometimes with minor motor features such as eye blinking or subtle chewing. The EEG finding in CAE is generalized 3 Hz spike and wave discharges. Some go on to develop generalized tonic-clonic seizures. This condition carries a good prognosis because children do not usually show cognitive decline or neurological deficits, and the seizures in the majority cease spontaneously with onging maturation.
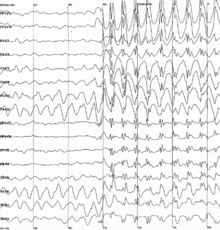
Generalized 3 Hz spike and wave discharges in EEG

- Juvenile absence epilepsy is an idiopathic generalized epilepsy with later onset that CAE, typically in prepubertal adolescence, with the most frequent seizure type being absence seizures. Generalized tonic-clonic seizures can occur. 3 Hz spike-wave or muliple spike discharges can be seen on EEG. Prognosis is mixed, with some patients going on to a syndrome that is poorly distinguishable from JME.
- Juvenile myoclonic epilepsy (JME) occurs in patients aged 8 to 20 years. Patients have normal cognition and are otherwise neurologically intact. The most common seizures are myoclonic jerks, although generalized tonic-clonic seizures and absence seizures may occur as well. Myoclonic jerks usually cluster in the early morning after awakening. The EEG reveals generalized 4-6 Hz spike wave discharges or multiple spike discharges. Interestingly, these patients are often first diagnosed when they have their first generalized tonic-clonic seizure later in life when they experience sleep deprivation (e.g., freshman year in college after staying up late to study for exams). Alcohol withdrawal can also be a major contributing factor in breakthrough seizures as well. The risk of the tendency to have seizures is lifelong; however, the majority have well-controlled seizures with anticonvulsant medication and avoidance of seizure precipitants.
- Symptomatic localization-related epilepsies Symptomatic localization-related epilepsies are divided by the location in the brain of the epileptic lesion, since the symptoms of the seizures are more closely tied to the brain location rather than the cause of the lesion. Tumors, atriovenous malformations, cavernous malformations, trauma, and cerebral infarcts can all be causes of epileptic foci in different brain regions.
- Temporal lobe epilepsy (TLE) is the most common epilepsy of adults who experience seizures poorly controlled with anticonvulsant medications. In most cases, the epileptogenic region is found in the midline (mesial) temporal structures (e.g., the hippocampus, amygdala, and parahippocampal gyrus). Seizures begin in late childhood and adolescence. Most of these patients have complex partial seizures sometimes preceded by an aura, and some TLE patients also suffer from secondary generalized tonic-clonic seizures. If the patient does not respond sufficiently to medical treatment, epilepsy surgery may be considered.
- Frontal lobe epilepsy arises from lesions causing seizures that occur in the frontal lobes of the brain. These epilepsies can be difficult to diagnose because the symptoms of seizures can easily be confused with nonepileptic spells and, because of limitations of the EEG, be difficult to "see" with standard scalp EEG.
- West syndrome is a triad of developmental delay, seizures termed infantile spasms, and EEG demonstrating a pattern termed hypsarrhythmia. Onset occurs between 3 months and 2 years, with peak onset between 8-9 months. West syndrome may arise from idiopathic, symptomatic, or cryptogenic causes. The most common cause is tuberous sclerosis. The prognosis varies with the underlying cause. In general most surviving patients remain with significant cognitive impairment and continuing seizures and may evolve to another eponymic syndrome, Lennox-Gastaut syndrome.
- Dravet's syndrome Severe myoclonic epilepsy of infancy (SMEI). This rare syndrome is distinguished from benign myoclonic epilepsy by its severity and must be differentiated from the Lennox-Gastaut syndrome and Doose’s myoclonic-astatic epilepsy. Onset is in the first year of life and symptoms peak at about 5 months of age with febrile hemiclonic or generalized status epilepticus. Boys are twice as often affected as girls. Prognosis is poor. Most cases are sporadic. Family history of epilepsy and febrile convulsions is present in around 25 percent of the cases.[5]
- Progressive myoclonic epilepsies define a group of symptomatic generalized epilepsies characterized by progressive dementia and myoclonic seizures. Tonic-clonic seizures may occur as well. Diseases usually classified in this group are Unverricht-Lundborg disease, myoclonus epilepsy with ragged red fibers, Lafora disease, neuronal ceroid lipofucinosis, and sialdosis.
- Lennox-Gastaut syndrome (LGS) is a triad of developmental delay or childhood dementia, mixed generalized seizures, and EEG demonstating a pattern of approximately 2 Hz "slow" spike-wave. Onset occurs between 2-18 years. As in West syndrome, LGS result from idiopathic, symptomatic, or cryptogenic causes, and many patients first have West syndrome. Authorities emphasize different seizure types as important in LGS, but most have astatic seizures (drop attacks), tonic seizures, tonic-clonic seizures, atypical absence seizures, and sometimes, complex partial seizures. Anticonvulsants are usually only partially successful in treatment.
References edit
- ^ Commission on Classification and Terminology of the International League Against Epilepsy (1989). "Proposal for revised classification of epilepsies and epileptic syndromes". Epilepsia. 30 (4): 389–99. doi:10.1111/j.1528-1157.1989.tb05316.x. PMID 2502382.
- ^ Loiseau P, Duché B, Cordova S, Dartigues JF, Cohadon S (1988). "Prognosis of benign childhood epilepsy with centrotemporal spikes: a follow-up study of 168 patients". Epilepsia. 29 (3): 229–35. doi:10.1111/j.1528-1157.1988.tb03711.x. PMID 3371279.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Panayiotopoulos CP (August 2000). "Benign childhood epileptic syndromes with occipital spikes: new classification proposed by the International League Against Epilepsy". J. Child Neurol. 15 (8): 548–52. doi:10.1177/088307380001500810. PMID 10961795.
{{cite journal}}: CS1 maint: date and year (link) - ^ Bertrand D, Picard F, Le Hellard S; et al. (2002). "How mutations in the nAChRs can cause ADNFLE epilepsy". Epilepsia. 43 (Suppl 5): 112–22. doi:10.1046/j.1528-1157.43.s.5.16.x. PMID 12121305.
{{cite journal}}: Explicit use of et al. in:|author=(help)CS1 maint: multiple names: authors list (link) - ^ "Dravet Syndrome". Retrieved 2007-12-16.