The urea cycle (also known as the ornithine cycle) is a cycle of biochemical reactions that produces urea (NH2)2CO from ammonia (NH3). Animals that use this cycle, mainly amphibians and mammals, are called ureotelic.
The urea cycle converts highly toxic ammonia to urea for excretion.[1] This cycle was the first metabolic cycle to be discovered (Hans Krebs and Kurt Henseleit, 1932), five years before the discovery of the TCA cycle. This cycle was described in more detail later on by Ratner and Cohen. The urea cycle takes place primarily in the liver and, to a lesser extent, in the kidneys.
Amino acid catabolism results in waste ammonia. All animals need a way to excrete this product. Most aquatic organisms, or ammonotelic organisms, excrete ammonia without converting it.[1] Organisms that cannot easily and safely remove nitrogen as ammonia convert it to a less toxic substance, such as urea, via the urea cycle, which occurs mainly in the liver. Urea produced by the liver is then released into the bloodstream, where it travels to the kidneys and is ultimately excreted in urine. The urea cycle is essential to these organisms, because if the nitrogen or ammonia is not eliminated from the organism it can be very detrimental.[2] In species including birds and most insects, the ammonia is converted into uric acid or its urate salt, which is excreted in solid form. Further, the urea cycle consumes acidic waste carbon dioxide by combining it with the basic ammonia, helping to maintain a neutral pH.
The entire process converts two amino groups, one from NH+ 4 and one from aspartate, and a carbon atom from HCO− 3, to the relatively nontoxic excretion product urea.[3] This occurs at the cost of four "high-energy" phosphate bonds (3 ATP hydrolyzed to 2 ADP and one AMP). The conversion from ammonia to urea happens in five main steps. The first is needed for ammonia to enter the cycle and the following four are all a part of the cycle itself. To enter the cycle, ammonia is converted to carbamoyl phosphate. The urea cycle consists of four enzymatic reactions: one mitochondrial and three cytosolic.[1][4] This uses 6 enzymes.[3][4][5]
Before the urea cycle begins ammonia is converted to carbamoyl phosphate. The reaction is catalyzed by carbamoyl phosphate synthetase I and requires the use of two ATP molecules.[1] The carbamoyl phosphate then enters the urea cycle.
Carbamoyl phosphate is converted to citrulline. With catalysis by ornithine transcarbamylase, the carbamoyl phosphate group is donated to ornithine and releases a phosphate group.[1]
Arginine is cleaved by arginase to form urea and ornithine. The ornithine is then transported back to the mitochondria to begin the urea cycle again.[1][4]
Since fumarate is obtained by removing NH3 from aspartate (by means of reactions 3 and 4), and PPi + H2O → 2 Pi, the equation can be simplified as follows:
Note that reactions related to the urea cycle also cause the production of 2 NADH, so the overall reaction releases slightly more energy than it consumes. The NADH is produced in two ways:
One NADH molecule is produced by the enzyme glutamate dehydrogenase in the conversion of glutamate to ammonium and α-ketoglutarate. Glutamate is the non-toxic carrier of amine groups. This provides the ammonium ion used in the initial synthesis of carbamoyl phosphate.
The fumarate released in the cytosol is hydrated to malate by cytosolic fumarase. This malate is then oxidized to oxaloacetate by cytosolic malate dehydrogenase, generating a reduced NADH in the cytosol. Oxaloacetate is one of the keto acids preferred by transaminases, and so will be recycled to aspartate, maintaining the flow of nitrogen into the urea cycle.
The two NADH produced can provide energy for the formation of 5 ATP (cytosolic NADH provides 2.5 ATP with the malate-aspartate shuttle in human liver cell), a net production of two high-energy phosphate bond for the urea cycle. However, if gluconeogenesis is underway in the cytosol, the latter reducing equivalent is used to drive the reversal of the GAPDH step instead of generating ATP.
The fate of oxaloacetate is either to produce aspartate via transamination or to be converted to phosphoenolpyruvate, which is a substrate for gluconeogenesis.
As stated above many vertebrates use the urea cycle to create urea out of ammonium so that the ammonium does not damage the body. Though this is helpful, there are other effects of the urea cycle. For example: consumption of two ATP, production of urea, generation of H+, the combining of HCO−3 and NH+4 to forms where it can be regenerated, and finally the consumption of NH+4.[6]
The synthesis of carbamoyl phosphate and the urea cycle are dependent on the presence of N-acetylglutamic acid (NAcGlu), which allosterically activates CPS1. NAcGlu is an obligate activator of carbamoyl phosphate synthetase.[7] Synthesis of NAcGlu by N-acetylglutamate synthase (NAGS) is stimulated by both Arg, allosteric stimulator of NAGS, and Glu, a product in the transamination reactions and one of NAGS's substrates, both of which are elevated when free amino acids are elevated. So Glu not only is a substrate for NAGS but also serves as an activator for the urea cycle.
The remaining enzymes of the cycle are controlled by the concentrations of their substrates. Thus, inherited deficiencies in cycle enzymes other than ARG1 do not result in significant decreases in urea production (if any cycle enzyme is entirely missing, death occurs shortly after birth). Rather, the deficient enzyme's substrate builds up, increasing the rate of the deficient reaction to normal.
The anomalous substrate buildup is not without cost, however. The substrate concentrations become elevated all the way back up the cycle to NH+ 4, resulting in hyperammonemia (elevated [NH+ 4]P).
Although the root cause of NH+ 4 toxicity is not completely understood, a high [NH+ 4] puts an enormous strain on the NH+ 4-clearing system, especially in the brain (symptoms of urea cycle enzyme deficiencies include intellectual disability and lethargy). This clearing system involves GLUD1 and GLUL, which decrease the 2-oxoglutarate (2OG) and Glu pools. The brain is most sensitive to the depletion of these pools. Depletion of 2OG decreases the rate of TCAC, whereas Glu is both a neurotransmitter and a precursor to GABA, another neurotransmitter. [1](p.734)
The urea cycle and the citric acid cycle are independent cycles but are linked. One of the nitrogen atoms in the urea cycle is obtained from the transamination of oxaloacetate to aspartate.[8] The fumarate that is produced in step three is also an intermediate in the citric acid cycle and is returned to that cycle.[8]
Urea cycle disorders are rare and affect about one in 35,000 people in the United States.[9]Genetic defects in the enzymes involved in the cycle can occur, which usually manifest within a few days after birth.[2] The recently born child will typically experience varying bouts of vomiting and periods of lethargy.[2] Ultimately, the infant may go into a coma and develop brain damage.[2] New-borns with UCD are at a much higher risk of complications or death due to untimely screening tests and misdiagnosed cases. The most common misdiagnosis is neonatal sepsis. Signs of UCD can be present within the first 2 to 3 days of life, but the present method to get confirmation by test results can take too long.[10] This can potentially cause complications such as coma or death.[10]
Urea cycle disorders may also be diagnosed in adults, and symptoms may include delirium episodes, lethargy, and symptoms similar to that of a stroke.[11] On top of these symptoms, if the urea cycle begins to malfunction in the liver, the patient may develop cirrhosis.[12] This can also lead to sarcopenia (the loss of muscle mass).[12] Mutations lead to deficiencies of the various enzymes and transporters involved in the urea cycle, and cause urea cycle disorders.[1] If individuals with a defect in any of the six enzymes used in the cycle ingest amino acids beyond what is necessary for the minimum daily requirements, then the ammonia that is produced will not be able to be converted to urea. These individuals can experience hyperammonemia, or the build-up of a cycle intermediate.
All urea cycle defects, except OTC deficiency, are inherited in an autosomal recessive manner. OTC deficiency is inherited as an X-linked recessive disorder, although some females can show symptoms. Most urea cycle disorders are associated with hyperammonemia, however argininemia and some forms of argininosuccinic aciduria do not present with elevated ammonia.
^ abcdTymoczko, John L.; Berg, Jeremy M.; Stryer, Lubert (2013). BIOCHEMISTRY A Short Course. W.H. Freeman and Company, New York. p. 529. ISBN978-1-4292-8360-1.
^Atkinson, Daniel (September 20, 1991). "Functional Roles of Urea in Vertebrates". Physiological Zoology. 65 (2) (2 ed.). Los Angeles: The University of Chicago Press: 243–267. doi:10.1086/physzool.65.2.30158252. JSTOR30158252. S2CID87121092.
^Kaplan Medical USMLE Step 1 Biochemistry and Medical Genetics Lecture Notes 2010, page 261
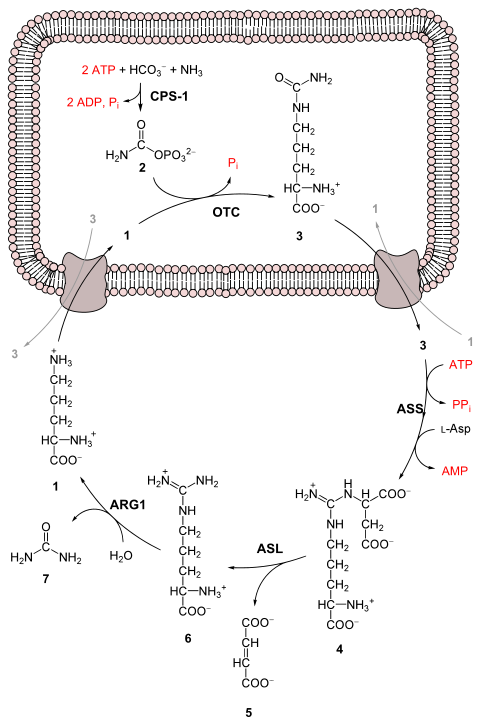

 Urea cycle.
Urea cycle. Urea cycle colored.
Urea cycle colored.
