T cells are one of the important types of white blood cells of the immune system and play a central role in the adaptive immune response. T cells can be distinguished from other lymphocytes by the presence of a T-cell receptor (TCR) on their cell surface.
| T cell | |
|---|---|
.png) Animation of T cell | |
 Scanning electron micrograph of a red blood cell (left), a platelet (center), and a T lymphocyte (right); colorized | |
| Details | |
| System | Immune system |
| Identifiers | |
| Latin | lymphocytus T |
| MeSH | D013601 |
| TH | H2.00.04.1.02007 |
| FMA | 62870 |
| Anatomical terms of microanatomy | |
T cells are born from hematopoietic stem cells,[1] found in the bone marrow. Developing T cells then migrate to the thymus gland to develop (or mature). T cells derive their name from the thymus.[2][3] After migration to the thymus, the precursor cells mature into several distinct types of T cells. T cell differentiation also continues after they have left the thymus. Groups of specific, differentiated T cell subtypes have a variety of important functions in controlling and shaping the immune response.
One of these functions is immune-mediated cell death, and it is carried out by two major subtypes: CD8+ "killer" (cytotoxic) and CD4+ "helper" T cells. (These are named for the presence of the cell surface proteins CD8 or CD4.) CD8+ T cells, also known as "killer T cells", are cytotoxic – this means that they are able to directly kill virus-infected cells, as well as cancer cells. CD8+ T cells are also able to use small signalling proteins, known as cytokines, to recruit other types of cells when mounting an immune response. A different population of T cells, the CD4+ T cells, function as "helper cells". Unlike CD8+ killer T cells, the CD4+ helper T (TH) cells function by further activating memory B cells and cytotoxic T cells, which leads to a larger immune response. The specific adaptive immune response regulated by the TH cell depends on its subtype (such as T-helper1, T-helper2, T-helper17, regulatory T-cell),[4] which is distinguished by the types of cytokines they secrete.[2]
Regulatory T cells are yet another distinct population of T cells that provide the critical mechanism of tolerance, whereby immune cells are able to distinguish invading cells from "self". This prevents immune cells from inappropriately reacting against one's own cells, known as an "autoimmune" response. For this reason, these regulatory T cells have also been called "suppressor" T cells. These same regulatory T cells can also be co-opted by cancer cells to prevent the recognition of, and an immune response against, tumor cells.
Development edit
Origin, early development and migration to the thymus edit
All T cells originate from c-kit+Sca1+ haematopoietic stem cells (HSC) which reside in the bone marrow. In some cases, the origin might be the fetal liver during embryonic development. The HSC then differentiate into multipotent progenitors (MPP) which retain the potential to become both myeloid and lymphoid cells. The process of differentiation then proceeds to a common lymphoid progenitor (CLP), which can only differentiate into T, B or NK cells.[5] These CLP cells then migrate via the blood to the thymus, where they engraft:. Henceforth they are known as thymocytes, the immature stage of a T cell.
The earliest cells which arrived in the thymus are commonly termed double-negative, as they express neither the CD4 nor CD8 co-receptor. The newly arrived CLP cells are CD4−CD8−CD44+CD25−ckit+ cells, and are termed early thymic progenitor (ETP) cells.[6] These cells will then undergo a round of division and downregulate c-kit and are termed double-negative one (DN1) cells. To become T cells, the thymocytes must undergo multiple DN stages as well as positive selection and negative selection.
Double negative thymocytes can be identified by the surface expression of CD2, CD5 and CD7. Still during the double negative stages, CD34 expression stops and CD1 is expressed. Expression of both CD4 and CD8 makes them double positive, and matures into either CD4+ or CD8+ cells.
TCR development edit
A critical step in T cell maturation is making a functional T cell receptor (TCR). Each mature T cell will ultimately contain a unique TCR that reacts to a random pattern, allowing the immune system to recognize many different types of pathogens. This process is essential in developing immunity to threats that the immune system has not encountered before, since due to random variation there will always be at least one TCR to match any new pathogen.
A thymocyte can only become an active T cell when it survives the process of developing a functional TCR. The TCR consists of two major components, the alpha and beta chains. These both contain random elements designed to produce a wide variety of different TCRs, but due to this huge variety they must be tested to make sure they work at all. First, the thymocytes attempt to create a functional beta chain, testing it against a 'mock' alpha chain. Then they attempt to create a functional alpha chain. Once a working TCR has been produced, the cells then must test if their TCR will identify threats correctly, and to do this it is required to recognize the body’s major histocompatibility complex (MHC) in a process known as positive selection. The thymocyte must also ensure that it does not react adversely to "self" antigens, called negative selection. If both positive and negative selection are successful, the TCR becomes fully operational and the thymocyte becomes a T cell.
TCR β-chain selection edit
At the DN2 stage (CD44+CD25+), cells upregulate the recombination genes RAG1 and RAG2 and re-arrange the TCRβ locus, combining V-D-J recombination and constant region genes in an attempt to create a functional TCRβ chain. As the developing thymocyte progresses through to the DN3 stage (CD44−CD25+), the thymocyte expresses an invariant α-chain called pre-Tα alongside the TCRβ gene. If the rearranged β-chain successfully pairs with the invariant α-chain, signals are produced which cease rearrangement of the β-chain (and silence the alternate allele).[7] Although these signals require the pre-TCR at the cell surface, they are independent of ligand binding to the pre-TCR. If the chains successfully pair a pre-TCR forms, and the cell downregulates CD25 and is termed a DN4 cell (CD25−CD44−). These cells then undergo a round of proliferation, and begin to re-arrange the TCRα locus during the double-positive stage.
Positive selection edit
The process of positive selection takes 3 to 4 days and occurs in the thymic cortex.[8] Double-positive thymocytes (CD4+/CD8+) migrate deep into the thymic cortex, where they are presented with self-antigens. These self-antigens are expressed by thymic cortical epithelial cells on MHC molecules, which reside on the surface of cortical epithelial cells. Only thymocytes that interact well with MHC-I or MHC-II will receive a vital "survival signal", while those that cannot interact strongly enough will receive no signal and die from neglect. This process ensures that the surviving thymocytes will have an 'MHC affinity' that means they can serve useful functions in the body, responding to MHC molecules to assist immune responses. The vast majority of developing thymocytes will not pass positive selection, and die during this process.[9]
A thymocyte's fate is determined during positive selection. Double-positive cells (CD4+/CD8+) that interact well with MHC class II molecules will eventually become CD4+ "helper" cells, whereas thymocytes that interact well with MHC class I molecules mature into CD8+ "killer" cells. A thymocyte becomes a CD4+ cell by down-regulating expression of its CD8 cell surface receptors. If the cell does not lose its signal, it will continue downregulating CD8 and become a CD4+, both CD8+ and CD4+ cells are now single positive cells.[10]
This process does not filter for thymocytes that may cause autoimmunity. The potentially autoimmune cells are removed by the following process of negative selection, which occurs in the thymic medulla.
Negative selection edit
Negative selection removes thymocytes that are capable of strongly binding with "self" MHC molecules. Thymocytes that survive positive selection migrate towards the boundary of the cortex and medulla in the thymus. While in the medulla, they are again presented with a self-antigen presented on the MHC complex of medullary thymic epithelial cells (mTECs).[11] mTECs must be Autoimmune regulator positive (AIRE+) to properly express self-antigens from all tissues of the body on their MHC class I peptides. Some mTECs are phagocytosed by thymic dendritic cells; this makes them AIRE− antigen presenting cells (APCs), allowing for presentation of self-antigens on MHC class II molecules (positively selected CD4+ cells must interact with these MHC class II molecules, thus APCs, which possess MHC class II, must be present for CD4+ T-cell negative selection). Thymocytes that interact too strongly with the self-antigen receive an apoptotic signal that leads to cell death. However, some of these cells are selected to become Treg cells. The remaining cells exit the thymus as mature naive T cells, also known as recent thymic emigrants.[12] This process is an important component of central tolerance and serves to prevent the formation of self-reactive T cells that are capable of inducing autoimmune diseases in the host.
TCR development summary edit
β-selection is the first checkpoint, where thymocytes that are able to form a functional pre-TCR (with an invariant alpha chain and a functional beta chain) are allowed to continue development in the thymus. Next, positive selection checks that thymocytes have successfully rearranged their TCRα locus and are capable of recognizing MHC molecules with appropriate affinity. Negative selection in the medulla then eliminates thymocytes that bind too strongly to self-antigens expressed on MHC molecules. These selection processes allow for tolerance of self by the immune system. Typical naive T cells that leave the thymus (via the corticomedullary junction) are self-restricted, self-tolerant, and single positive.
Thymic output edit
About 98% of thymocytes die during the development processes in the thymus by failing either positive selection or negative selection, whereas the other 2% survive and leave the thymus to become mature immunocompetent T cells.[13] The thymus contributes fewer cells as a person ages. As the thymus shrinks by about 3%[14] a year throughout middle age, a corresponding fall in the thymic production of naive T cells occurs, leaving peripheral T cell expansion and regeneration to play a greater role in protecting older people.
Types of T cell edit
T cells are grouped into a series of subsets based on their function. CD4 and CD8 T cells are selected in the thymus, but undergo further differentiation in the periphery to specialized cells which have different functions. T cell subsets were initially defined by function, but also have associated gene or protein expression patterns.
Conventional adaptive T cells edit
Helper CD4+ T cells edit
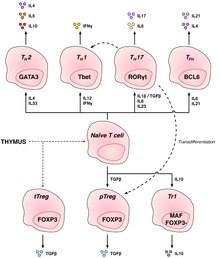
T helper cells (TH cells) assist other lymphocytes, including the maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages. These cells are also known as CD4+ T cells as they express the CD4 glycoprotein on their surfaces. Helper T cells become activated when they are presented with peptide antigens by MHC class II molecules, which are expressed on the surface of antigen-presenting cells (APCs). Once activated, they divide rapidly and secrete cytokines that regulate or assist the immune response. These cells can differentiate into one of several subtypes, which have different roles. Cytokines direct T cells into particular subtypes.[15]
| Cell type | Cytokines Produced | Key Transcription Factor | Role in immune defense | Related diseases |
|---|---|---|---|---|
| Th1 | IFNγ, IL-2 | Tbet | Produce an inflammatory response, key for defense against intracellular bacteria, viruses and cancer. | MS, Type 1 diabetes |
| Th2 | IL-4, IL-5, IL-13 | GATA-3 | Immunologically important against extracellular pathogens, such as worm infections | Asthma and other allergic diseases |
| Th17 | IL-17F, IL-17A, IL-22 | RORγt | Defense against gut pathogens and at mucosal barriers | MS, Rheumatoid Arthritis, Psoriasis |
| Th9[16][17] | IL-9 | IRF4, PU.1 | Defense against helminths (parasitic worms) and cell-dependent allergic inflammation | Multiple Sclerosis |
| Tfh | IL-21, IL-4 | Bcl-6 | Help B cells produce antibodies | Asthma and other allergic diseases |
| Th22[18][17] | IL-22 | AHR | Pathogenesis of allergic airway diseases and predominantly anti-inflammatory | Crohn's Disease, Rheumatoid Arthritis, Tumors |
Cytotoxic CD8+ T cells edit

Cytotoxic T cells (TC cells, CTLs, T-killer cells, killer T cells) destroy virus-infected cells and tumor cells, and are also implicated in transplant rejection. These cells are defined by the expression of the CD8 protein on their cell surface. Cytotoxic T cells recognize their targets by binding to short peptides (8-11 amino acids in length) associated with MHC class I molecules, present on the surface of all nucleated cells. Cytotoxic T cells also produce the key cytokines IL-2 and IFNγ. These cytokines influence the effector functions of other cells, in particular macrophages and NK cells.
Memory T cells edit
Antigen-naive T cells expand and differentiate into memory and effector T cells after they encounter their cognate antigen within the context of an MHC molecule on the surface of a professional antigen presenting cell (e.g. a dendritic cell). Appropriate co-stimulation must be present at the time of antigen encounter for this process to occur. Historically, memory T cells were thought to belong to either the effector or central memory subtypes, each with their own distinguishing set of cell surface markers (see below).[19] Subsequently, numerous new populations of memory T cells were discovered including tissue-resident memory T (Trm) cells, stem memory TSCM cells, and virtual memory T cells. The single unifying theme for all memory T cell subtypes is that they are long-lived and can quickly expand to large numbers of effector T cells upon re-exposure to their cognate antigen. By this mechanism they provide the immune system with "memory" against previously encountered pathogens. Memory T cells may be either CD4+ or CD8+ and usually express CD45RO.[20]
Memory T cell subtypes:
- Central memory T cells (TCM cells) express CD45RO, C-C chemokine receptor type 7 (CCR7), and L-selectin (CD62L). Central memory T cells also have intermediate to high expression of CD44. This memory subpopulation is commonly found in the lymph nodes and in the peripheral circulation. (Note- CD44 expression is usually used to distinguish murine naive from memory T cells).
- Effector memory T cells (TEM cells and TEMRA cells) express CD45RO but lack expression of CCR7 and L-selectin. They also have intermediate to high expression of CD44. These memory T cells lack lymph node-homing receptors and are thus found in the peripheral circulation and tissues.[21] TEMRA stands for terminally differentiated effector memory cells re-expressing CD45RA, which is a marker usually found on naive T cells.[22]
- Tissue-resident memory T cells (TRM) occupy tissues (skin, lung, etc.) without recirculating. One cell surface marker that has been associated with TRM is the intern αeβ7, also known as CD103.[23]
- Virtual memory T cells (TVM) differ from the other memory subsets in that they do not originate following a strong clonal expansion event. Thus, although this population as a whole is abundant within the peripheral circulation, individual virtual memory T cell clones reside at relatively low frequencies. One theory is that homeostatic proliferation gives rise to this T cell population. Although CD8 virtual memory T cells were the first to be described,[24] it is now known that CD4 virtual memory cells also exist.[25]
Regulatory CD4+ T cells edit
Regulatory T cells are crucial for the maintenance of immunological tolerance. Their major role is to shut down T cell–mediated immunity toward the end of an immune reaction and to suppress autoreactive T cells that escaped the process of negative selection in the thymus.
Two major classes of CD4+ Treg cells have been described—FOXP3+ Treg cells and FOXP3− Treg cells.
Regulatory T cells can develop either during normal development in the thymus, and are then known as thymic Treg cells, or can be induced peripherally and are called peripherally derived Treg cells. These two subsets were previously called "naturally occurring" and "adaptive" (or "induced"), respectively.[26] Both subsets require the expression of the transcription factor FOXP3 which can be used to identify the cells. Mutations of the FOXP3 gene can prevent regulatory T cell development, causing the fatal autoimmune disease IPEX.
Several other types of T cells have suppressive activity, but do not express FOXP3 constitutively. These include Tr1 and Th3 cells, which are thought to originate during an immune response and act by producing suppressive molecules. Tr1 cells are associated with IL-10, and Th3 cells are associated with TGF-beta. Recently, Th17 cells have been added to this list.[27]
Innate-like T cells edit
Innate-like T cells or unconventional T cells represent some subsets of T cells that behave differently in immunity. They trigger rapid immune responses, regardless of the major histocompatibility complex (MHC) expression, unlike their conventional counterparts (CD4 T helper cells and CD8 cytotoxic T cells), which are dependent on the recognition of peptide antigens in the context of the MHC molecule. Overall, there are three large populations of unconventional T cells: NKT cells, MAIT cells, and gammadelta T cells. Now, their functional roles are already being well established in the context of infections and cancer.[28] Furthermore, these T cell subsets are being translated into many therapies against malignancies such as leukemia, for example.[29]
Natural killer T cell edit
Natural killer T cells (NKT cells – not to be confused with natural killer cells of the innate immune system) bridge the adaptive immune system with the innate immune system. Unlike conventional T cells that recognize protein peptide antigens presented by major histocompatibility complex (MHC) molecules, NKT cells recognize glycolipid antigens presented by CD1d. Once activated, these cells can perform functions ascribed to both helper and cytotoxic T cells: cytokine production and release of cytolytic/cell killing molecules. They are also able to recognize and eliminate some tumor cells and cells infected with herpes viruses.[30]
Mucosal associated invariant T cells edit
Mucosal associated invariant T (MAIT) cells display innate, effector-like qualities.[31][32] In humans, MAIT cells are found in the blood, liver, lungs, and mucosa, defending against microbial activity and infection.[31] The MHC class I-like protein, MR1, is responsible for presenting bacterially-produced vitamin B metabolites to MAIT cells.[33][34][35] After the presentation of foreign antigen by MR1, MAIT cells secrete pro-inflammatory cytokines and are capable of lysing bacterially-infected cells.[31][35] MAIT cells can also be activated through MR1-independent signaling.[35] In addition to possessing innate-like functions, this T cell subset supports the adaptive immune response and has a memory-like phenotype.[31] Furthermore, MAIT cells are thought to play a role in autoimmune diseases, such as multiple sclerosis, arthritis and inflammatory bowel disease,[36][37] although definitive evidence is yet to be published.[38][39][40][41]
Gamma delta T cells edit
Gamma delta T cells (γδ T cells) represent a small subset of T cells which possess a γδ TCR rather than the αβ TCR on the cell surface. The majority of T cells express αβ TCR chains. This group of T cells is much less common in humans and mice (about 2% of total T cells) and are found mostly in the gut mucosa, within a population of intraepithelial lymphocytes. In rabbits, sheep, and chickens, the number of γδ T cells can be as high as 60% of total T cells. The antigenic molecules that activate γδ T cells are still mostly unknown. However, γδ T cells are not MHC-restricted and seem to be able to recognize whole proteins rather than requiring peptides to be presented by MHC molecules on APCs. Some murine γδ T cells recognize MHC class IB molecules. Human γδ T cells that use the Vγ9 and Vδ2 gene fragments constitute the major γδ T cell population in peripheral blood. These cells are unique in that they specifically and rapidly respond to a set of nonpeptidic phosphorylated isoprenoid precursors, collectively named phosphoantigens, which are produced by virtually all living cells. The most common phosphoantigens from animal and human cells (including cancer cells) are isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMPP). Many microbes produce the active compound hydroxy-DMAPP (HMB-PP) and corresponding mononucleotide conjugates, in addition to IPP and DMAPP. Plant cells produce both types of phosphoantigens. Drugs activating human Vγ9/Vδ2 T cells comprise synthetic phosphoantigens and aminobisphosphonates, which upregulate endogenous IPP/DMAPP.
Activation edit

Activation of CD4+ T cells occurs through the simultaneous engagement of the T-cell receptor and a co-stimulatory molecule (like CD28, or ICOS) on the T cell by the major histocompatibility complex (MHCII) peptide and co-stimulatory molecules on the APC. Both are required for production of an effective immune response; in the absence of co-stimulation, T cell receptor signalling alone results in anergy. The signalling pathways downstream from co-stimulatory molecules usually engages the PI3K pathway generating PIP3 at the plasma membrane and recruiting PH domain containing signaling molecules like PDK1 that are essential for the activation of PKC-θ, and eventual IL-2 production. Optimal CD8+ T cell response relies on CD4+ signalling.[43] CD4+ cells are useful in the initial antigenic activation of naive CD8 T cells, and sustaining memory CD8+ T cells in the aftermath of an acute infection. Therefore, activation of CD4+ T cells can be beneficial to the action of CD8+ T cells.[44][45][46]
The first signal is provided by binding of the T cell receptor to its cognate peptide presented on MHCII on an APC. MHCII is restricted to so-called professional antigen-presenting cells, like dendritic cells, B cells, and macrophages, to name a few. The peptides presented to CD8+ T cells by MHC class I molecules are 8–13 amino acids in length; the peptides presented to CD4+ cells by MHC class II molecules are longer, usually 12–25 amino acids in length,[47] as the ends of the binding cleft of the MHC class II molecule are open.
The second signal comes from co-stimulation, in which surface receptors on the APC are induced by a relatively small number of stimuli, usually products of pathogens, but sometimes breakdown products of cells, such as necrotic-bodies or heat shock proteins. The only co-stimulatory receptor expressed constitutively by naive T cells is CD28, so co-stimulation for these cells comes from the CD80 and CD86 proteins, which together constitute the B7 protein, (B7.1 and B7.2, respectively) on the APC. Other receptors are expressed upon activation of the T cell, such as OX40 and ICOS, but these largely depend upon CD28 for their expression. The second signal licenses the T cell to respond to an antigen. Without it, the T cell becomes anergic, and it becomes more difficult for it to activate in future. This mechanism prevents inappropriate responses to self, as self-peptides will not usually be presented with suitable co-stimulation. Once a T cell has been appropriately activated (i.e. has received signal one and signal two) it alters its cell surface expression of a variety of proteins. Markers of T cell activation include CD69, CD71 and CD25 (also a marker for Treg cells), and HLA-DR (a marker of human T cell activation). CTLA-4 expression is also up-regulated on activated T cells, which in turn outcompetes CD28 for binding to the B7 proteins. This is a checkpoint mechanism to prevent over activation of the T cell. Activated T cells also change their cell surface glycosylation profile.[48]
The T cell receptor exists as a complex of several proteins. The actual T cell receptor is composed of two separate peptide chains, which are produced from the independent T cell receptor alpha and beta (TCRα and TCRβ) genes. The other proteins in the complex are the CD3 proteins: CD3εγ and CD3εδ heterodimers and, most important, a CD3ζ homodimer, which has a total of six ITAM motifs. The ITAM motifs on the CD3ζ can be phosphorylated by Lck and in turn recruit ZAP-70. Lck and/or ZAP-70 can also phosphorylate the tyrosines on many other molecules, not least CD28, LAT and SLP-76, which allows the aggregation of signalling complexes around these proteins.
Phosphorylated LAT recruits SLP-76 to the membrane, where it can then bring in PLC-γ, VAV1, Itk and potentially PI3K. PLC-γ cleaves PI(4,5)P2 on the inner leaflet of the membrane to create the active intermediaries diacylglycerol (DAG), inositol-1,4,5-trisphosphate (IP3); PI3K also acts on PIP2, phosphorylating it to produce phosphatidlyinositol-3,4,5-trisphosphate (PIP3). DAG binds and activates some PKCs. Most important in T cells is PKC-θ, critical for activating the transcription factors NF-κB and AP-1. IP3 is released from the membrane by PLC-γ and diffuses rapidly to activate calcium channel receptors on the ER, which induces the release of calcium into the cytosol. Low calcium in the endoplasmic reticulum causes STIM1 clustering on the ER membrane and leads to activation of cell membrane CRAC channels that allows additional calcium to flow into the cytosol from the extracellular space. This aggregated cytosolic calcium binds calmodulin, which can then activate calcineurin. Calcineurin, in turn, activates NFAT, which then translocates to the nucleus. NFAT is a transcription factor that activates the transcription of a pleiotropic set of genes, most notable, IL-2, a cytokine that promotes long-term proliferation of activated T cells.
PLC-γ can also initiate the NF-κB pathway. DAG activates PKC-θ, which then phosphorylates CARMA1, causing it to unfold and function as a scaffold. The cytosolic domains bind an adapter BCL10 via CARD (Caspase activation and recruitment domains) domains; that then binds TRAF6, which is ubiquitinated at K63.: 513–523 [49] This form of ubiquitination does not lead to degradation of target proteins. Rather, it serves to recruit NEMO, IKKα and -β, and TAB1-2/ TAK1.[50] TAK 1 phosphorylates IKK-β, which then phosphorylates IκB allowing for K48 ubiquitination: leads to proteasomal degradation. Rel A and p50 can then enter the nucleus and bind the NF-κB response element. This coupled with NFAT signaling allows for complete activation of the IL-2 gene.[49]
While in most cases activation is dependent on TCR recognition of antigen, alternative pathways for activation have been described. For example, cytotoxic T cells have been shown to become activated when targeted by other CD8 T cells leading to tolerization of the latter.[51]
In spring 2014, the T-Cell Activation in Space (TCAS) experiment was launched to the International Space Station on the SpaceX CRS-3 mission to study how "deficiencies in the human immune system are affected by a microgravity environment".[52]
T cell activation is modulated by reactive oxygen species.[53]
Antigen discrimination edit
A unique feature of T cells is their ability to discriminate between healthy and abnormal (e.g. infected or cancerous) cells in the body.[54] Healthy cells typically express a large number of self derived pMHC on their cell surface and although the T cell antigen receptor can interact with at least a subset of these self pMHC, the T cell generally ignores these healthy cells. However, when these very same cells contain even minute quantities of pathogen derived pMHC, T cells are able to become activated and initiate immune responses. The ability of T cells to ignore healthy cells but respond when these same cells contain pathogen (or cancer) derived pMHC is known as antigen discrimination. The molecular mechanisms that underlie this process are controversial.[54][55]
Clinical significance edit
Deficiency edit
.jpg)
Causes of T cell deficiency include lymphocytopenia of T cells and/or defects on function of individual T cells. Complete insufficiency of T cell function can result from hereditary conditions such as severe combined immunodeficiency (SCID), Omenn syndrome, and cartilage–hair hypoplasia.[56] Causes of partial insufficiencies of T cell function include acquired immune deficiency syndrome (AIDS), and hereditary conditions such as DiGeorge syndrome (DGS), chromosomal breakage syndromes (CBSs), and B cell and T cell combined disorders such as ataxia-telangiectasia (AT) and Wiskott–Aldrich syndrome (WAS).[56]
The main pathogens of concern in T cell deficiencies are intracellular pathogens, including Herpes simplex virus, Mycobacterium and Listeria.[57] Also, fungal infections are also more common and severe in T cell deficiencies.[57]
Cancer edit
Cancer of T cells is termed T-cell lymphoma, and accounts for perhaps one in ten cases of non-Hodgkin lymphoma.[58] The main forms of T cell lymphoma are:
- Extranodal T cell lymphoma
- Cutaneous T cell lymphomas: Sézary syndrome and Mycosis fungoides
- Anaplastic large cell lymphoma
- Angioimmunoblastic T cell lymphoma
Exhaustion edit
It has been suggested that this section be split out into another article titled T cell exhaustion. (Discuss) (May 2023) |
T cell exhaustion is a poorly defined or ambiguous term.[59][60] There are three approaches to its definition.[59] "The first approach primarily defines as exhausted the cells that present the same cellular dysfunction (typically, the absence of an expected effector response). The second approach primarily defines as exhausted the cells that are produced by a given cause (typically, but not necessarily, chronic exposure to an antigen). Finally, the third approach primarily defines as exhausted the cells that present the same molecular markers (typically, programmed cell death protein 1 [PD-1])."[59]
Dysfunctional T cells are characterized by progressive loss of function, changes in transcriptional profiles and sustained expression of inhibitory receptors. At first, cells lose their ability to produce IL-2 and TNFα, which is followed by the loss of high proliferative capacity and cytotoxic potential, and eventually leads to their deletion. Exhausted T cells typically indicate higher levels of CD43, CD69 and inhibitory receptors combined with lower expression of CD62L and CD127. Exhaustion can develop during chronic infections, sepsis and cancer.[61] Exhausted T cells preserve their functional exhaustion even after repeated antigen exposure.[62]
During chronic infection and sepsis edit
T cell exhaustion can be triggered by several factors like persistent antigen exposure and lack of CD4 T cell help.[63] Antigen exposure also has effect on the course of exhaustion because longer exposure time and higher viral load increases the severity of T cell exhaustion. At least 2–4 weeks exposure is needed to establish exhaustion.[64] Another factor able to induce exhaustion are inhibitory receptors including programmed cell death protein 1 (PD1), CTLA-4, T cell membrane protein-3 (TIM3), and lymphocyte activation gene 3 protein (LAG3).[65][66] Soluble molecules such as cytokines IL-10 or TGF-β are also able to trigger exhaustion.[67][68] Last known factors that can play a role in T cell exhaustion are regulatory cells. Treg cells can be a source of IL-10 and TGF-β and therefore they can play a role in T cell exhaustion.[69] Furthermore, T cell exhaustion is reverted after depletion of Treg cells and blockade of PD1.[70] T cell exhaustion can also occur during sepsis as a result of cytokine storm. Later after the initial septic encounter anti-inflammatory cytokines and pro-apoptotic proteins take over to protect the body from damage. Sepsis also carries high antigen load and inflammation. In this stage of sepsis T cell exhaustion increases.[71][72] Currently there are studies aiming to utilize inhibitory receptor blockades in treatment of sepsis.[73][74][75]
During transplantation edit
While during infection T cell exhaustion can develop following persistent antigen exposure after graft transplant similar situation arises with alloantigen presence.[76] It was shown that T cell response diminishes over time after kidney transplant.[77] These data suggest T cell exhaustion plays an important role in tolerance of a graft mainly by depletion of alloreactive CD8 T cells.[72][78] Several studies showed positive effect of chronic infection on graft acceptance and its long-term survival mediated partly by T cell exhaustion.[79][80][81] It was also shown that recipient T cell exhaustion provides sufficient conditions for NK cell transfer.[82] While there are data showing that induction of T cell exhaustion can be beneficial for transplantation it also carries disadvantages among which can be counted increased number of infections and the risk of tumor development.[83]
During cancer edit
During cancer T cell exhaustion plays a role in tumor protection. According to research some cancer-associated cells as well as tumor cells themselves can actively induce T cell exhaustion at the site of tumor.[84][85][86] T cell exhaustion can also play a role in cancer relapses as was shown on leukemia.[87] Some studies have suggested that it is possible to predict relapse of leukemia based on expression of inhibitory receptors PD-1 and TIM-3 by T cells.[88] Many experiments and clinical trials have focused on immune checkpoint blockers in cancer therapy, with some of these approved as valid therapies that are now in clinical use.[89] Inhibitory receptors targeted by those medical procedures are vital in T cell exhaustion and blocking them can reverse these changes.[90]
See also edit
References edit
- ^ "5. Hematopoietic Stem Cells". Stem Cell Information. Bethesda, MD: National Institutes of Health, U.S. Department of Health and Human Services. 17 June 2001. Archived from the original on 29 October 2016. Retrieved 21 December 2021.
- ^ a b Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002). "Helper T Cells and Lymphocyte Activation". Molecular Biology of the Cell (4th ed.). Garland Science.
- ^ Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002). "Helper t Cells and Lymphocyte Activation". Molecular Biology of the Cell (4th ed.). Garland Science. p. 1367.
T cells ... derive their [name] from the organs in which they develop. T cells develop [mature] in the thymus
- ^ Luckheeram RV, Zhou R, Verma AD, Xia B (2012). "CD4⁺T cells: differentiation and functions". Clinical & Developmental Immunology. 2012: 925135. doi:10.1155/2012/925135. PMC 3312336. PMID 22474485.
- ^ Kondo M (December 2016). "One Niche to Rule Both Maintenance and Loss of Stemness in HSCs". Immunity. 45 (6): 1177–1179. doi:10.1016/j.immuni.2016.12.003. PMID 28002722.
- ^ Osborne LC, Dhanji S, Snow JW, Priatel JJ, Ma MC, Miners MJ, et al. (March 2007). "Impaired CD8 T cell memory and CD4 T cell primary responses in IL-7R alpha mutant mice". The Journal of Experimental Medicine. 204 (3): 619–631. doi:10.1084/jem.20061871. PMC 2137912. PMID 17325202.
- ^ Murphy, Kenneth (2011). Janeway's Immunobiology (8th ed.). Garland Science. pp. 301–305. ISBN 9780815342434.
- ^ Ross JO, Melichar HJ, Au-Yeung BB, Herzmark P, Weiss A, Robey EA (June 2014). "Distinct phases in the positive selection of CD8+ T cells distinguished by intrathymic migration and T-cell receptor signaling patterns". Proceedings of the National Academy of Sciences of the United States of America. 111 (25): E2550–E2558. Bibcode:2014PNAS..111E2550R. doi:10.1073/pnas.1408482111. PMC 4078834. PMID 24927565.
- ^ Starr TK, Jameson SC, Hogquist KA (2003-01-01). "Positive and negative selection of T cells". Annual Review of Immunology. 21 (1): 139–176. doi:10.1146/annurev.immunol.21.120601.141107. PMID 12414722.
- ^ Zerrahn J, Held W, Raulet DH (March 1997). "The MHC reactivity of the T cell repertoire prior to positive and negative selection". Cell. 88 (5): 627–636. doi:10.1016/S0092-8674(00)81905-4. PMID 9054502. S2CID 15983629.
- ^ Hinterberger M, Aichinger M, Prazeres da Costa O, Voehringer D, Hoffmann R, Klein L (June 2010). "Autonomous role of medullary thymic epithelial cells in central CD4(+) T cell tolerance" (PDF). Nature Immunology. 11 (6): 512–519. doi:10.1038/ni.1874. PMID 20431619. S2CID 33154019.
- ^ Pekalski ML, García AR, Ferreira RC, Rainbow DB, Smyth DJ, Mashar M, Brady J, Savinykh N, Dopico XC, Mahmood S, Duley S, Stevens HE, Walker NM, Cutler AJ, Waldron-Lynch F, Dunger DB, Shannon-Lowe C, Coles AJ, Jones JL, Wallace C, Todd JA, Wicker LS (August 2017). "Neonatal and adult recent thymic emigrants produce IL-8 and express complement receptors CR1 and CR2". JCI Insight. 2 (16). doi:10.1172/jci.insight.93739. PMC 5621870. PMID 28814669.
- ^ Murphy, Kenneth (2011). Janeway's Immunobiology (8th ed.). Garland Science. p. 297. ISBN 9780815342434.
- ^ Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP (2000). "The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection". Annu. Rev. Immunol. 18: 529–560. doi:10.1146/annurev.immunol.18.1.529. PMID 10837068.
- ^ Gutcher I, Becher B (2007). "APC-derived cytokines and T cell polarization in autoimmune inflammation". J. Clin. Invest. 117 (5): 1119–27. doi:10.1172/JCI31720. PMC 1857272. PMID 17476341.
- ^ Wang W, Sung N, Gilman-Sachs A, Kwak-Kim J (18 August 2020). "T Helper (Th) Cell Profiles in Pregnancy and Recurrent Pregnancy Losses: Th1/Th2/Th9/Th17/Th22/Tfh Cells". Frontiers in Immunology. 11: 2025. doi:10.3389/fimmu.2020.02025. PMC 7461801. PMID 32973809.
- ^ a b Saravia J, Chapman NM, Chi H (July 2019). "Helper T cell differentiation". Cellular & Molecular Immunology. 16 (7): 634–643. doi:10.1038/s41423-019-0220-6. PMC 6804569. PMID 30867582.
- ^ Jia L, Wu C (2014). "The Biology and Functions of Th22 Cells". T Helper Cell Differentiation and Their Function. Advances in Experimental Medicine and Biology. Vol. 841. pp. 209–230. doi:10.1007/978-94-017-9487-9_8. ISBN 978-94-017-9486-2. PMID 25261209.
- ^ Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A (1999). "Two subsets of memory T lymphocytes with distinct homing potentials and effector functions". Nature. 401 (6754): 708–712. Bibcode:1999Natur.401..708S. doi:10.1038/44385. PMID 10537110. S2CID 4378970.
- ^ Akbar AN, Terry L, Timms A, Beverley PC, Janossy G (April 1988). "Loss of CD45R and gain of UCHL1 reactivity is a feature of primed T cells". J. Immunol. 140 (7): 2171–8. doi:10.4049/jimmunol.140.7.2171. PMID 2965180. S2CID 22340282.
- ^ Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF (2005). "Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets" (PDF). Journal of Immunology. 175 (9): 5895–903. doi:10.4049/jimmunol.175.9.5895. PMID 16237082. S2CID 16412760.
- ^ Koch S, Larbi A, Derhovanessian E, Özcelik D, Naumova E, Pawelec G (2008). "Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people". Immunity & Ageing. 5 (6): 6. doi:10.1186/1742-4933-5-6. PMC 2515281. PMID 18657274.
- ^ Shin H, Iwasaki A (September 2013). "Tissue-resident memory T cells". Immunological Reviews. 255 (1): 165–81. doi:10.1111/imr.12087. PMC 3748618. PMID 23947354.
- ^ Lee YJ, Jameson SC, Hogquist KA (2011). "Alternative memory in the CD8 T cell lineage". Trends in Immunology. 32 (2): 50–56. doi:10.1016/j.it.2010.12.004. PMC 3039080. PMID 21288770.
- ^ Marusina AI, Ono Y, Merleev AA, Shimoda M, Ogawa H, Wang EA, Kondo K, Olney L, Luxardi G, Miyamura Y, Yilma TD, Villalobos IB, Bergstrom JW, Kronenberg DG, Soulika AM, Adamopoulos IE, Maverakis E (2017). "CD4+ virtual memory: Antigen-inexperienced T cells reside in the naïve, regulatory, and memory T cell compartments at similar frequencies, implications for autoimmunity". Journal of Autoimmunity. 77: 76–88. doi:10.1016/j.jaut.2016.11.001. PMC 6066671. PMID 27894837.
- ^ Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, Jiang S, Kuchroo VK, Mathis D, Roncarolo MG, Rudensky A, Sakaguchi S, Shevach EM, Vignali DA, Ziegler SF (2013). "Regulatory T cells: recommendations to simplify the nomenclature". Nat. Immunol. 14 (4): 307–8. doi:10.1038/ni.2554. PMID 23507634. S2CID 11294516.
- ^ Singh B, Schwartz JA, Sandrock C, Bellemore SM, Nikoopour E (2013). "Modulation of autoimmune diseases by interleukin (IL)-17 producing regulatory T helper (Th17) cells". Indian J. Med. Res. 138 (5): 591–4. PMC 3928692. PMID 24434314.
- ^ Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB (November 2015). "The burgeoning family of unconventional T cells". Nature Immunology. 16 (11): 1114–1123. doi:10.1038/ni.3298. PMID 26482978. S2CID 30992456.
- ^ de Araújo ND, Gama FM, de Souza Barros M, Ribeiro TL, Alves FS, Xabregas LA, et al. (2021). "Translating Unconventional T Cells and Their Roles in Leukemia Antitumor Immunity". Journal of Immunology Research. 2021: 6633824. doi:10.1155/2021/6633824. PMC 7808823. PMID 33506055.
- ^ Mallevaey T, Fontaine J, Breuilh L, Paget C, Castro-Keller A, Vendeville C, Capron M, Leite-de-Moraes M, Trottein F, Faveeuw C (May 2007). "Invariant and noninvariant natural killer T cells exert opposite regulatory functions on the immune response during murine schistosomiasis". Infection and Immunity. 75 (5): 2171–80. doi:10.1128/IAI.01178-06. PMC 1865739. PMID 17353286.
- ^ a b c d Napier RJ, Adams EJ, Gold MC, Lewinsohn DM (2015-07-06). "The Role of Mucosal Associated Invariant T Cells in Antimicrobial Immunity". Frontiers in Immunology. 6: 344. doi:10.3389/fimmu.2015.00344. PMC 4492155. PMID 26217338.
- ^ Gold MC, Lewinsohn DM (August 2011). "Mucosal associated invariant T cells and the immune response to infection". Microbes and Infection. 13 (8–9): 742–8. doi:10.1016/j.micinf.2011.03.007. PMC 3130845. PMID 21458588.
- ^ Eckle SB, Corbett AJ, Keller AN, Chen Z, Godfrey DI, Liu L, Mak JY, Fairlie DP, Rossjohn J, McCluskey J (December 2015). "Recognition of Vitamin B Precursors and Byproducts by Mucosal Associated Invariant T Cells". The Journal of Biological Chemistry. 290 (51): 30204–11. doi:10.1074/jbc.R115.685990. PMC 4683245. PMID 26468291.
- ^ Ussher JE, Klenerman P, Willberg CB (2014-10-08). "Mucosal-associated invariant T-cells: new players in anti-bacterial immunity". Frontiers in Immunology. 5: 450. doi:10.3389/fimmu.2014.00450. PMC 4189401. PMID 25339949.
- ^ a b c Howson LJ, Salio M, Cerundolo V (2015-06-16). "MR1-Restricted Mucosal-Associated Invariant T Cells and Their Activation during Infectious Diseases". Frontiers in Immunology. 6: 303. doi:10.3389/fimmu.2015.00303. PMC 4468870. PMID 26136743.
- ^ Hinks TS (May 2016). "Mucosal-associated invariant T cells in autoimmunity, immune-mediated diseases and airways disease". Immunology. 148 (1): 1–12. doi:10.1111/imm.12582. PMC 4819138. PMID 26778581.
- ^ Bianchini E, De Biasi S, Simone AM, Ferraro D, Sola P, Cossarizza A, Pinti M (March 2017). "Invariant natural killer T cells and mucosal-associated invariant T cells in multiple sclerosis". Immunology Letters. 183: 1–7. doi:10.1016/j.imlet.2017.01.009. PMID 28119072.
- ^ Serriari NE, Eoche M, Lamotte L, Lion J, Fumery M, Marcelo P, Chatelain D, Barre A, Nguyen-Khac E, Lantz O, Dupas JL, Treiner E (May 2014). "Innate mucosal-associated invariant T (MAIT) cells are activated in inflammatory bowel diseases". Clinical and Experimental Immunology. 176 (2): 266–74. doi:10.1111/cei.12277. PMC 3992039. PMID 24450998.
- ^ Huang S, Martin E, Kim S, Yu L, Soudais C, Fremont DH, Lantz O, Hansen TH (May 2009). "MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution". Proceedings of the National Academy of Sciences of the United States of America. 106 (20): 8290–5. Bibcode:2009PNAS..106.8290H. doi:10.1073/pnas.0903196106. PMC 2688861. PMID 19416870.
- ^ Chua WJ, Hansen TH (November 2010). "Bacteria, mucosal-associated invariant T cells and MR1". Immunology and Cell Biology. 88 (8): 767–9. doi:10.1038/icb.2010.104. PMID 20733595. S2CID 27717815.
- ^ Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, Bhati M, Chen Z, Kostenko L, Reantragoon R, Williamson NA, Purcell AW, Dudek NL, McConville MJ, O'Hair RA, Khairallah GN, Godfrey DI, Fairlie DP, Rossjohn J, McCluskey J (November 2012). "MR1 presents microbial vitamin B metabolites to MAIT cells" (PDF). Nature. 491 (7426): 717–23. Bibcode:2012Natur.491..717K. doi:10.1038/nature11605. PMID 23051753. S2CID 4419703.
- ^ The NIAID resource booklet "Understanding the Immune System (pdf)".
- ^ Williams MA, Bevan MJ (2007-01-01). "Effector and memory CTL differentiation". Annual Review of Immunology. 25 (1): 171–92. doi:10.1146/annurev.immunol.25.022106.141548. PMID 17129182.
- ^ Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP (February 2003). "CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes". Nature. 421 (6925): 852–6. Bibcode:2003Natur.421..852J. doi:10.1038/nature01441. PMID 12594515. S2CID 574770.
- ^ Shedlock DJ, Shen H (April 2003). "Requirement for CD4 T cell help in generating functional CD8 T cell memory". Science. 300 (5617): 337–9. Bibcode:2003Sci...300..337S. doi:10.1126/science.1082305. PMID 12690201. S2CID 38040377.
- ^ Sun JC, Williams MA, Bevan MJ (September 2004). "CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection". Nature Immunology. 5 (9): 927–33. doi:10.1038/ni1105. PMC 2776074. PMID 15300249.
- ^ Jennifer Rolland and Robyn O'Hehir, "Turning off the T cells: Peptides for treatment of allergic Diseases," Today's life science publishing, 1999, Page 32
- ^ Maverakis E, Kim K, Shimoda M, Gershwin M, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB (2015). "Glycans in the immune system and The Altered Glycan Theory of Autoimmunity". J Autoimmun. 57 (6): 1–13. doi:10.1016/j.jaut.2014.12.002. PMC 4340844. PMID 25578468.
- ^ a b Tatham P, Gomperts BD, Kramer IM (2003). Signal transduction. Amsterdam: Elsevier Academic Press. ISBN 978-0-12-289632-3.
- ^ Wu H, Arron JR (November 2003). "TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology". BioEssays. 25 (11): 1096–105. doi:10.1002/bies.10352. PMID 14579250. S2CID 28521713.
- ^ Milstein O, Hagin D, Lask A, Reich-Zeliger S, Shezen E, Ophir E, Eidelstein Y, Afik R, Antebi YE, Dustin ML, Reisner Y (January 2011). "CTLs respond with activation and granule secretion when serving as targets for T cell recognition". Blood. 117 (3): 1042–52. doi:10.1182/blood-2010-05-283770. PMC 3035066. PMID 21045195.
- ^ Graham W (2014-04-14). "SpaceX ready for CRS-3 Dragon launch and new milestones". NASAspaceflight.com. Retrieved 2014-04-14.
- ^ Belikov AV, Schraven B, Simeoni L (October 2015). "T cells and reactive oxygen species". Journal of Biomedical Science. 22: 85. doi:10.1186/s12929-015-0194-3. PMC 4608155. PMID 26471060.
- ^ a b Feinerman O, Germain RN, Altan-Bonnet G (2008). "Quantitative challenges in understanding ligand discrimination by alphabeta T cells". Mol. Immunol. 45 (3): 619–31. doi:10.1016/j.molimm.2007.03.028. PMC 2131735. PMID 17825415.
- ^ Dushek O, van der Merwe PA (2014). "An induced rebinding model of antigen discrimination". Trends Immunol. 35 (4): 153–8. doi:10.1016/j.it.2014.02.002. PMC 3989030. PMID 24636916.
- ^ a b T-Cell Disorders at eMedicine
- ^ a b Jones J, Bannister BA, Gillespie SH, eds. (2006). Infection: Microbiology and Management. Wiley-Blackwell. p. 435. ISBN 978-1-4051-2665-6.
- ^ "The Lymphomas" (PDF). The Leukemia & Lymphoma Society. May 2006. p. 2. Retrieved 2008-04-07.
- ^ a b c Kaminski H, Lemoine M, Pradeu T (September 2021). "Immunological exhaustion: How to make a disparate concept operational?". PLOS Pathogens. 17 (9): e1009892. doi:10.1371/journal.ppat.1009892. PMC 8460019. PMID 34555119.
- ^ Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. (November 2019). "Defining 'T cell exhaustion'". Nature Reviews. Immunology. 19 (11): 665–674. doi:10.1038/s41577-019-0221-9. PMC 7286441. PMID 31570879.
- ^ Yi JS, Cox MA, Zajac AJ (April 2010). "T-cell exhaustion: characteristics, causes and conversion". Immunology. 129 (4): 474–81. doi:10.1111/j.1365-2567.2010.03255.x. PMC 2842494. PMID 20201977.
- ^ Wang Q, Pan W, Liu Y, Luo J, Zhu D, Lu Y, Feng X, Yang X, Dittmer U, Lu M, Yang D, Liu J (2018). "Hepatitis B Virus-Specific CD8+ T Cells Maintain Functional Exhaustion after Antigen Reexposure in an Acute Activation Immune Environment". Front Immunol. 9: 219. doi:10.3389/fimmu.2018.00219. PMC 5816053. PMID 29483916.
- ^ Matloubian M, Concepcion RJ, Ahmed R (December 1994). "CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection". Journal of Virology. 68 (12): 8056–63. doi:10.1128/JVI.68.12.8056-8063.1994. PMC 237269. PMID 7966595.
- ^ Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ (August 2012). "Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection". Journal of Virology. 86 (15): 8161–70. doi:10.1128/JVI.00889-12. PMC 3421680. PMID 22623779.
- ^ Wherry EJ (June 2011). "T cell exhaustion". Nature Immunology. 12 (6): 492–9. doi:10.1038/ni.2035. PMID 21739672. S2CID 11052693.
- ^ Okagawa T, Konnai S, Nishimori A, Maekawa N, Goto S, Ikebuchi R, Kohara J, Suzuki Y, Yamada S, Kato Y, Murata S, Ohashi K (June 2018). "+ T cells during bovine leukemia virus infection". Veterinary Research. 49 (1): 50. doi:10.1186/s13567-018-0543-9. PMC 6006750. PMID 29914540.
- ^ Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB (November 2006). "Interleukin-10 determines viral clearance or persistence in vivo". Nature Medicine. 12 (11): 1301–9. doi:10.1038/nm1492. PMC 2535582. PMID 17041596.
- ^ Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI (July 2009). "Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo". Immunity. 31 (1): 145–57. doi:10.1016/j.immuni.2009.06.015. PMC 3039716. PMID 19604493.
- ^ Veiga-Parga T, Sehrawat S, Rouse BT (September 2013). "Role of regulatory T cells during virus infection". Immunological Reviews. 255 (1): 182–96. doi:10.1111/imr.12085. PMC 3748387. PMID 23947355.
- ^ Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE, O'Mara L, Yang S, Konieczny BT, Sharpe AH, Freeman GJ, Rudensky AY, Ahmed R (August 2014). "Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection". The Journal of Experimental Medicine. 211 (9): 1905–18. doi:10.1084/jem.20132577. PMC 4144726. PMID 25113973.
- ^ Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, Bauer M, Riedemann NC (July 2011). "The late phase of sepsis is characterized by an increased microbiological burden and death rate". Critical Care. 15 (4): R183. doi:10.1186/cc10332. PMC 3387626. PMID 21798063.
- ^ a b Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS (December 2011). "Immunosuppression in patients who die of sepsis and multiple organ failure". JAMA. 306 (23): 2594–605. doi:10.1001/jama.2011.1829. PMC 3361243. PMID 22187279.
- ^ Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS (February 2017). "Anti-PD-L1 peptide improves survival in sepsis". The Journal of Surgical Research. 208: 33–39. doi:10.1016/j.jss.2016.08.099. PMC 5535083. PMID 27993215.
- ^ Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS (December 2016). "Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1". Journal of Leukocyte Biology. 100 (6): 1239–1254. doi:10.1189/jlb.4hi0616-255r. PMC 5110001. PMID 27671246.
- ^ Wei Z, Li P, Yao Y, Deng H, Yi S, Zhang C, Wu H, Xie X, Xia M, He R, Yang XP, Tang ZH (July 2018). "Alpha-lactose reverses liver injury via blockade of Tim-3-mediated CD8 apoptosis in sepsis". Clinical Immunology. 192: 78–84. doi:10.1016/j.clim.2018.04.010. PMID 29689313. S2CID 21657071.
- ^ Wells AD, Li XC, Strom TB, Turka LA (May 2001). "The role of peripheral T-cell deletion in transplantation tolerance". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 356 (1409): 617–23. doi:10.1098/rstb.2001.0845. PMC 1088449. PMID 11375065.
- ^ Halloran PF, Chang J, Famulski K, Hidalgo LG, Salazar ID, Merino Lopez M, Matas A, Picton M, de Freitas D, Bromberg J, Serón D, Sellarés J, Einecke G, Reeve J (July 2015). "Disappearance of T Cell-Mediated Rejection Despite Continued Antibody-Mediated Rejection in Late Kidney Transplant Recipients". Journal of the American Society of Nephrology. 26 (7): 1711–20. doi:10.1681/ASN.2014060588. PMC 4483591. PMID 25377077.
- ^ Steger U, Denecke C, Sawitzki B, Karim M, Jones ND, Wood KJ (May 2008). "Exhaustive differentiation of alloreactive CD8+ T cells: critical for determination of graft acceptance or rejection" (PDF). Transplantation. 85 (9): 1339–47. doi:10.1097/TP.0b013e31816dd64a. PMID 18475193. S2CID 33409478.
- ^ de Mare-Bredemeijer EL, Shi XL, Mancham S, van Gent R, van der Heide-Mulder M, de Boer R, Heemskerk MH, de Jonge J, van der Laan LJ, Metselaar HJ, Kwekkeboom J (August 2015). "Cytomegalovirus-Induced Expression of CD244 after Liver Transplantation Is Associated with CD8+ T Cell Hyporesponsiveness to Alloantigen". Journal of Immunology. 195 (4): 1838–48. doi:10.4049/jimmunol.1500440. PMID 26170387.
- ^ Gassa A, Jian F, Kalkavan H, Duhan V, Honke N, Shaabani N, Friedrich SK, Dolff S, Wahlers T, Kribben A, Hardt C, Lang PA, Witzke O, Lang KS (2016). "IL-10 Induces T Cell Exhaustion During Transplantation of Virus Infected Hearts". Cellular Physiology and Biochemistry. 38 (3): 1171–81. doi:10.1159/000443067. PMID 26963287.
- ^ Shi XL, de Mare-Bredemeijer EL, Tapirdamaz Ö, Hansen BE, van Gent R, van Campenhout MJ, Mancham S, Litjens NH, Betjes MG, van der Eijk AA, Xia Q, van der Laan LJ, de Jonge J, Metselaar HJ, Kwekkeboom J (September 2015). "CMV Primary Infection Is Associated With Donor-Specific T Cell Hyporesponsiveness and Fewer Late Acute Rejections After Liver Transplantation". American Journal of Transplantation. 15 (9): 2431–42. doi:10.1111/ajt.13288. PMID 25943855. S2CID 5348557.
- ^ Williams RL, Cooley S, Bachanova V, Blazar BR, Weisdorf DJ, Miller JS, Verneris MR (March 2018). "Recipient T Cell Exhaustion and Successful Adoptive Transfer of Haploidentical Natural Killer Cells". Biology of Blood and Marrow Transplantation. 24 (3): 618–622. doi:10.1016/j.bbmt.2017.11.022. PMC 5826878. PMID 29197679.
- ^ Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA (February 2012). "Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape". Cancer Research. 72 (4): 917–27. doi:10.1158/0008-5472.CAN-11-1620. PMC 3288154. PMID 22186141.
- ^ Zelle-Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, et al. (November 2016). "T cells in multiple myeloma display features of exhaustion and senescence at the tumor site". Journal of Hematology & Oncology. 9 (1): 116. doi:10.1186/s13045-016-0345-3. PMC 5093947. PMID 27809856.
- ^ Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD (March 2018). "Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells". Nature Communications. 9 (1): 948. Bibcode:2018NatCo...9..948L. doi:10.1038/s41467-018-03347-0. PMC 5838096. PMID 29507342.
- ^ Conforti L (February 2012). "The ion channel network in T lymphocytes, a target for immunotherapy". Clinical Immunology. 142 (2): 105–106. doi:10.1016/j.clim.2011.11.009. PMID 22189042.
- ^ Liu L, Chang YJ, Xu LP, Zhang XH, Wang Y, Liu KY, Huang XJ (May 2018). "T cell exhaustion characterized by compromised MHC class I and II restricted cytotoxic activity associates with acute B lymphoblastic leukemia relapse after allogeneic hematopoietic stem cell transplantation". Clinical Immunology. 190: 32–40. doi:10.1016/j.clim.2018.02.009. PMID 29477343.
- ^ Kong Y, Zhang J, Claxton DF, Ehmann WC, Rybka WB, Zhu L, et al. (July 2015). "PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation". Blood Cancer Journal. 5 (7): e330. doi:10.1038/bcj.2015.58. PMC 4526784. PMID 26230954.
- ^ "U.S. FDA Approved Immune-Checkpoint Inhibitors and Immunotherapies". Medical Writer Agency | 香港醫學作家 | MediPR | MediPaper Hong Kong. 2018-08-21. Retrieved 2018-09-22.
- ^ Bhadra R, Gigley JP, Weiss LM, Khan IA (May 2011). "Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade". Proceedings of the National Academy of Sciences of the United States of America. 108 (22): 9196–9201. Bibcode:2011PNAS..108.9196B. doi:10.1073/pnas.1015298108. PMC 3107287. PMID 21576466.
Further reading edit
- Janeway Jr CA, Travers P, Walport M, Shlomchik MJ (2001). Immunobiology 5 : the immune system in health and disease (5th ed.). New York: Garland Science. ISBN 978-0-8153-3642-6.
- "The Immune System" (PDF). National Institute of Allergy and Infectious Diseases. September 2003. Archived from the original (PDF) on 25 June 2009.